Community hub
Abortive initiation
View on Wikipedia
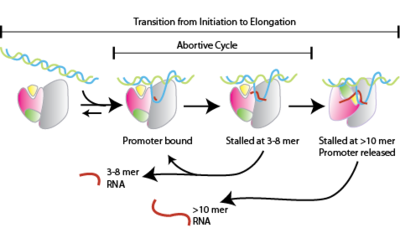
Abortive initiation, also known as abortive transcription, is an early process of genetic transcription in which RNA polymerase binds to a DNA promoter and enters into cycles of synthesis of short mRNA transcripts which are released before the transcription complex leaves the promoter. This process occurs in both eukaryotes and prokaryotes. Abortive initiation is typically studied in the T3 and T7 RNA polymerases in bacteriophages and in E. coli.
Overall process
[edit]Abortive initiation occurs prior to promoter clearance.[1]
- RNA polymerase binds to promoter DNA to form an RNA polymerase-promoter closed complex
- RNA polymerase then unwinds one turn of DNA surrounding the transcription start site to yield an RNA polymerase-promoter open complex
- RNA polymerase enters into abortive cycles of synthesis and releases short RNA products (contains less than 10 nucleotides)
- RNA polymerase escapes the promoter and enters into the elongation step of transcription
Mechanism
[edit]Abortive initiation is a normal process of transcription and occurs both in vitro and in vivo.[2] After each nucleotide-addition step in initial transcription, RNA polymerase, stochastically, can proceed on the pathway toward promoter escape (productive initiation) or can release the RNA product and revert to the RNA polymerase-promoter open complex (abortive initiation). During this early stage of transcription, RNA polymerase enters a phase during which dissociation of the transcription complex energetically competes with the elongation process. Abortive cycling is not caused by strong binding between the initiation complex and the promoter.[3]
DNA scrunching
[edit]
For many years, the mechanism by which RNA polymerase moves along the DNA strand during abortive initiation remained elusive. It had been observed that RNA polymerase did not escape from the promoter during transcription initiation, so it was unknown how the enzyme could read the DNA strand to transcribe it without moving downstream. Within the last decade, studies have revealed that abortive initiation involves DNA scrunching, in which RNA polymerase remains stationary while it unwinds and pulls downstream DNA into the transcription complex to pass the nucleotides through the polymerase active site, thereby transcribing the DNA without moving. This causes the unwound DNA to accumulate within the enzyme, hence the name DNA "scrunching". In abortive initiation, RNA polymerase re-winds and ejects the downstream portion of the unwound DNA, releasing the RNA, and reverting to the RNA polymerase-promoter open complex; in contrast, in productive initiation, RNA polymerase re-winds and ejects the upstream portion of the unwound DNA, breaking RNA polymerase-promoter interactions, escaping the promoter, and forming a transcription elongation complex.[1][4]
A 2006 paper that demonstrated the involvement of DNA scrunching in initial transcription proposed the idea that the stress incurred during DNA scrunching provides the driving force for both abortive initiation and productive initiation.[4] A companion paper published the same year confirmed that detectable DNA scrunching occurs in 80% of transcription cycles, and is actually estimated to be 100%, given the limitation of the ability to detect rapid scrunching (20% of scrunches have a duration of less than 1 second).[1]
A 2016 paper showed that DNA scrunching also occurs before RNA synthesis during transcription start site selection.[5]
Function
[edit]There are no widely accepted functions for the resulting truncated RNA transcripts. However, a study in 1981 found evidence that there was a relationship between the number of abortive transcripts produced and the time until long RNA strands are successfully produced. When RNA polymerase undergoes abortive transcription in the presence of ATP, UTP, and GTP, a complex is formed that has a much lower capacity for abortive recycling and a much higher rate of synthesis of the full-length RNA transcript.[6] A study in 2010 found evidence that these truncated transcripts inhibit termination of RNA synthesis by a RNA hairpin-dependent intrinsic terminator.[7]
See also
[edit]References
[edit]- ^ a b c Revyakin A, Liu C, Ebright RH, Strick TR (2006). "Abortive initiation and productive initiation by RNA polymerase involve DNA scrunching". Science. 314 (5802): 1139–43. Bibcode:2006Sci...314.1139R. doi:10.1126/science.1131398. PMC 2754787. PMID 17110577.
- ^ Goldman S, Ebright RH, Nickels B (2009). "Direct detection of abortive RNA transcripts in vivo". Science. 324 (5929): 927–928. Bibcode:2009Sci...324..927G. doi:10.1126/science.1169237. PMC 2718712. PMID 19443781.
- ^ Martin CT, Muller DK, Coleman JE (1988). "Processivity in early stages of transcription by T7 RNA polymerase". Biochemistry. 27 (11): 3966–74. doi:10.1021/bi00411a012. PMID 3415967.
- ^ a b Kapanidis AN, Margeat E, Ho SO, Kortkhonjia E, Weiss S, Ebright RH (2006). "Initial transcription by RNA polymerase proceeds through a DNA-scrunching mechanism". Science. 314 (5802): 1144–7. Bibcode:2006Sci...314.1144K. doi:10.1126/science.1131399. PMC 2754788. PMID 17110578.
- ^ Winkelman JT, Vvedenskaya IO, Zhang Y, Zhang Y, Bird JG, Taylor DM, Gourse RL, Ebright RH, Nickels BE (2016). "Multiplexed protein-DNA cross-linking: Scrunching in transcription start site selection". Science. 351 (6277): 1090–3. Bibcode:2016Sci...351.1090W. doi:10.1126/science.aad6881. PMC 4797950. PMID 26941320.
- ^ Munson LM, Reznikoff WS (1981). "Abortive initiation and long ribonucleic acid synthesis". Biochemistry. 20 (8): 2081–5. doi:10.1021/bi00511a003. PMID 6165380.
- ^ Lee S, Nguyen HM, Kang C (2010). "Tiny abortive initiation transcripts exert antitermination activity on an RNA hairpin-dependent intrinsic terminator". Nucleic Acids Res. 38 (18): 6045–53. doi:10.1093/nar/gkq450. PMC 2952870. PMID 20507918.