
Community hub
Recent from talks
Contribute something to knowledge base
Content stats: 0 posts, 0 articles, 1 media, 0 notes
Members stats: 0 subscribers, 0 contributors, 0 moderators, 0 supporters
Subscribers
Supporters
Contributors
Moderators
Hub AI
MELAS syndrome AI simulator
(@MELAS syndrome_simulator)
Hub AI
MELAS syndrome AI simulator
(@MELAS syndrome_simulator)
MELAS syndrome
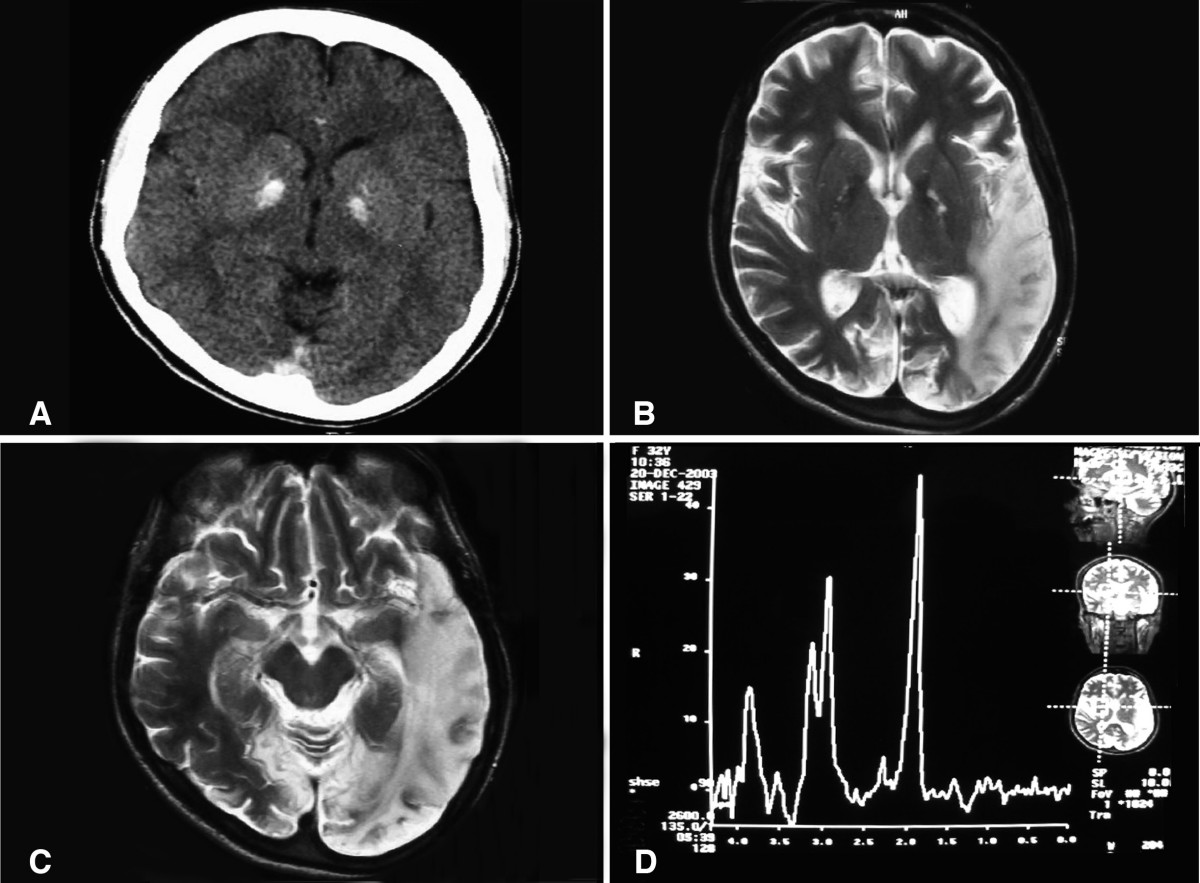
MELAS (Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like episodes) is one of the family of mitochondrial diseases, which also include MIDD (maternally inherited diabetes and deafness), MERRF syndrome, and Leber's hereditary optic neuropathy. It was first characterized under this name in 1984. A feature of these diseases is that they are caused by defects in the mitochondrial genome which is inherited purely from the female parent. The most common MELAS mutation is one in mitochondrial DNA (mtDNA) referred to as m.3243A>G.
MELAS is a condition that affects many of the body's systems, particularly the brain and nervous system (encephalo-) and muscles (myopathy). As such, it is commonly referred to as a mitochondrial encephalomyopathy, due to the co-occurrence of these pathologies. In most cases, the signs and symptoms of this disorder appear in childhood following a period of normal development. Children with MELAS often have normal early psychomotor development until the onset of symptoms between 2 and 10 years old. Though less common, infantile onset may occur and may present as failure to thrive, growth retardation and progressive deafness. Onset in older children typically presents as recurrent attacks of a migraine-like headache, anorexia, vomiting, and seizures. Children with MELAS are also frequently found to have short stature.
Most people with MELAS have a buildup of lactic acid in their bodies, a condition called lactic acidemia. Increased acidity in the blood can lead to vomiting, abdominal pain, extreme tiredness (fatigue), muscle weakness, loss of bowel control, and difficulty breathing. Less commonly, people with MELAS may experience involuntary muscle spasms (myoclonus), impaired muscle coordination (ataxia), hearing loss, heart and kidney problems, diabetes, epilepsy, and hormonal imbalances. Lactic acidemia also promotes mitochondrial dysfunction, one of the hallmarks of MELAS pathophysiology.
The presentation of some cases is similar to that of Kearns–Sayre syndrome.
Myoclonus epilepsy associated with ragged red fibers (MERRF) may be confused with MELAS as they both involve seizures, mental deterioration, and myopathy with ragged red fibers on biopsy. MERRF patients may also have hearing loss, visual disturbance secondary to optic atrophy, and short stature. The characteristic myoclonic seizure in MERRF may help to narrow diagnosis, but genetic testing should be considered to distinguish the two conditions.
Leigh syndrome may also present with progressive neurological deterioration, seizures, and vomiting, mainly in young children.
MELAS is mostly caused by mutations in the genes in mitochondrial DNA, but it can also be caused by mutations in the nuclear DNA.
Some of the genes (MT-ND1, MT-ND5) affected in MELAS encode proteins that are part of NADH dehydrogenase (also called complex I) in mitochondria, that helps convert oxygen and simple sugars to energy.
MELAS syndrome
MELAS (Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like episodes) is one of the family of mitochondrial diseases, which also include MIDD (maternally inherited diabetes and deafness), MERRF syndrome, and Leber's hereditary optic neuropathy. It was first characterized under this name in 1984. A feature of these diseases is that they are caused by defects in the mitochondrial genome which is inherited purely from the female parent. The most common MELAS mutation is one in mitochondrial DNA (mtDNA) referred to as m.3243A>G.
MELAS is a condition that affects many of the body's systems, particularly the brain and nervous system (encephalo-) and muscles (myopathy). As such, it is commonly referred to as a mitochondrial encephalomyopathy, due to the co-occurrence of these pathologies. In most cases, the signs and symptoms of this disorder appear in childhood following a period of normal development. Children with MELAS often have normal early psychomotor development until the onset of symptoms between 2 and 10 years old. Though less common, infantile onset may occur and may present as failure to thrive, growth retardation and progressive deafness. Onset in older children typically presents as recurrent attacks of a migraine-like headache, anorexia, vomiting, and seizures. Children with MELAS are also frequently found to have short stature.
Most people with MELAS have a buildup of lactic acid in their bodies, a condition called lactic acidemia. Increased acidity in the blood can lead to vomiting, abdominal pain, extreme tiredness (fatigue), muscle weakness, loss of bowel control, and difficulty breathing. Less commonly, people with MELAS may experience involuntary muscle spasms (myoclonus), impaired muscle coordination (ataxia), hearing loss, heart and kidney problems, diabetes, epilepsy, and hormonal imbalances. Lactic acidemia also promotes mitochondrial dysfunction, one of the hallmarks of MELAS pathophysiology.
The presentation of some cases is similar to that of Kearns–Sayre syndrome.
Myoclonus epilepsy associated with ragged red fibers (MERRF) may be confused with MELAS as they both involve seizures, mental deterioration, and myopathy with ragged red fibers on biopsy. MERRF patients may also have hearing loss, visual disturbance secondary to optic atrophy, and short stature. The characteristic myoclonic seizure in MERRF may help to narrow diagnosis, but genetic testing should be considered to distinguish the two conditions.
Leigh syndrome may also present with progressive neurological deterioration, seizures, and vomiting, mainly in young children.
MELAS is mostly caused by mutations in the genes in mitochondrial DNA, but it can also be caused by mutations in the nuclear DNA.
Some of the genes (MT-ND1, MT-ND5) affected in MELAS encode proteins that are part of NADH dehydrogenase (also called complex I) in mitochondria, that helps convert oxygen and simple sugars to energy.
Recent media
Recent media