Community hub
Recent from talks
Knowledge base stats:
Talk channels stats:
Members stats:
Ollier disease
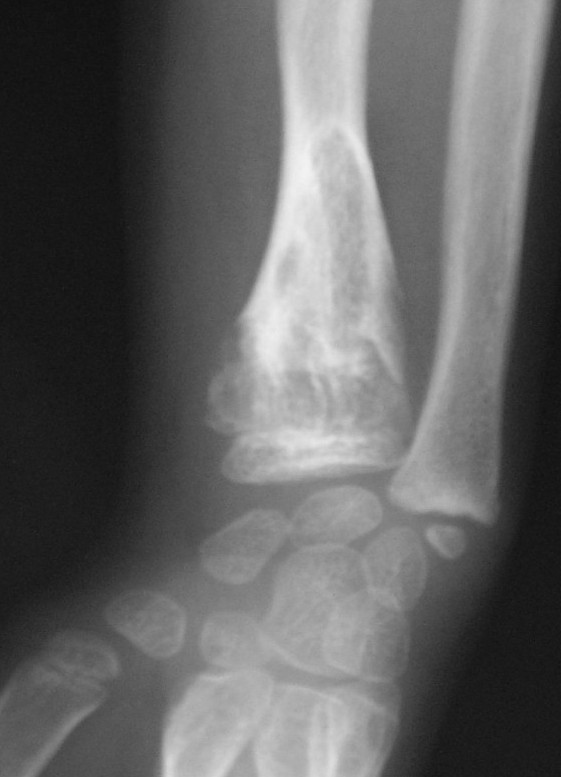
Ollier disease is a rare sporadic nonhereditary skeletal disorder in which typically benign cartilaginous tumors (enchondromas) develop near the growth plate cartilage. This is caused by cartilage rests that grow and reside within the metaphysis or diaphysis and eventually mineralize over time to form multiple enchondromas. Key signs of the disorder include asymmetry and shortening of the limb as well as an increased thickness of the bone margin. These symptoms are typically first visible during early childhood with the mean age of diagnosis being 13 years of age. Many patients with Ollier disease are prone to develop other malignancies including bone sarcomas that necessitate treatment and the removal of malignant bone neoplasm. Cases in patients with Ollier disease has shown a link to IDH1, IDH2, and PTH1R gene mutations. Currently, there are no forms of treatment for the underlying condition of Ollier disease but complications such as fractures, deformities, malignancies that arise from it can be treated through surgical procedures. The prevalence of this condition is estimated at around 1 in 100,000. It is unclear whether the men or women are more affected by this disorder due to conflicting case studies.
The disease consists of the growth multiple enchondromas which usually develop in early childhood. The growth of these enchondromas usually stops after skeletal maturation. The affected extremity is shortened (asymmetric dwarfism) and sometimes bowed due to epiphyseal fusion anomalies. Bone lesions generally present as cellular during childhood and become more solitary over time. People with Ollier disease are prone to breaking bones (fractures) and normally have swollen, aching limbs. However, many cases of solitary enchondromata go unnoticed due to lack of symptoms. Enchondromas are commonly found in the phalanges, metacarpal, and metatarsal bones in patients of Ollier disease due to the affinity of enchondromas to long tubular bones such as the femur and humerus. A unilateral distribution of bone lesions is usually observed but bilateral distributions or a singular extremity can occur as well. Approximately a third of the cases show some form of physical deformities of bowing or abnormal limb lengthening.[citation needed]
Ollier disease carries a higher risk of malignancies such as central nervous system (CNS), ovarian, and adenocarcinoma. Cranial gliomas have been linked with this disorder at an increased rate and at an earlier diagnosis age. A majority of glioma cases contain IDH gene mutations thus explaining the link between the two conditions. Juvenile granulosa cell tumour has also been associated with the disease. One case study indicates that this is due a mesodermal dysplasia in the long bones resulting in ovarian cancer.
The incidence of a secondary chondrosarcoma in Ollier disease is most commonly approximated at 25–30% with some projections even as high as 50%. Chondrosarcomas are typically developed during young adulthood and mostly form as a unifocal distribution in Ollier disease patients. The most common locations of tumors are in the pelvis and shoulder girdle. While chondrosarcoma is the most common form of a secondary malignant bone neoplasm found in cases of Ollier disease, other forms such as chordomas and osteosarcomas can occur. If left untreated, these malignant transformations may lead to fatal outcomes.[citation needed]
A related and even rarer disorder named Maffucci syndrome is a very similar condition that is characterized by the presence of multiple enchondromas with hemangiomas and occasionally lymphangiomas usually near the hands and feet but not limited to the skull, ribs, and spinal bones. This disorder is also sporadic and nonhereditary and usually detected during childhood despite being a congenital condition. Maffucci syndrome has also similarly been linked to IDH1 and IDH2 mutations specifically the IDH1 R132C hotspot mutation. This hotspot mutation is presumed to be responsible for the spindle cell hemangiomas and enchondromas in cases of Maffucci syndrome. Sanger sequencing analysis concluded that exon 4 is the primary location of mutations in IDH1 and IDH2 genes are specifically responsible for hemangiomas. Maffucci syndrome carries a significantly higher risk of malignant transformations like chondrosarcomas but also much more aggressive tumors such as acute lymphocytic leukemia and gastrointestinal and ovarian malignancies.
For many years, most research has been inconclusive regarding the cause of the disease.
Recent studies have shown that most cases of Ollier disease are believed to have been caused by isocitrate dehydrogenases IDH1 and IDH2 mutations. In one study, 35 of 43 (81%) patients with Ollier disease had either a IDH1 or IDH2 mutation. Another study suggests that R132C IDH1 mutations which are particularly dominant at exon 4 of IDH genes are linked to the growth of vascular lesions. Isocitrate deyhydrogenases IDH1 and IDH2 are catalysts responsible for the conversion of isocitrate to 2-oxoglutarate. The isocitrate deyhydrogenases IDH1 and IDH2 mutations disrupt this process resulting in unregulated production of α-ketoglutarate and a reduction in chondrocyte proliferation. In many cases of cartilaginous tumors, IDH1 and IDH2 point mutations were found thus explaining why Ollier disease is associated with many different associated conditions. Based on these case studies, most evidence suggests that the abnormal lining of lesions found in Ollier disease would suggest that the condition is caused by a post-zygotic somatic mutation thus resulting in a mosaic genetic disorder.
Approximately 8–10% of cases of patients with Ollier disease have been linked to PTH1R mutations. A particular case study the mutant heterozygous PTHR1 (R150C) receptor was observed in two unrelated patients with Ollier disease. This PTHR1 (R150C) mutant causes a reduction in chondrocyte differentiation by triggering the PTHrP-dependent pathway and decreasing PTHLH receptor function by approximately 30% creating enchondromas. One of these patients with the PTHR1 (R150C) mutant was found to have inherited the mutation from his father. This provides credence to the theory that multiple genetic mutations are needed to occur in order for Ollier disease to manifest.
Hub AI
Ollier disease AI simulator
(@Ollier disease_simulator)
Ollier disease
Ollier disease is a rare sporadic nonhereditary skeletal disorder in which typically benign cartilaginous tumors (enchondromas) develop near the growth plate cartilage. This is caused by cartilage rests that grow and reside within the metaphysis or diaphysis and eventually mineralize over time to form multiple enchondromas. Key signs of the disorder include asymmetry and shortening of the limb as well as an increased thickness of the bone margin. These symptoms are typically first visible during early childhood with the mean age of diagnosis being 13 years of age. Many patients with Ollier disease are prone to develop other malignancies including bone sarcomas that necessitate treatment and the removal of malignant bone neoplasm. Cases in patients with Ollier disease has shown a link to IDH1, IDH2, and PTH1R gene mutations. Currently, there are no forms of treatment for the underlying condition of Ollier disease but complications such as fractures, deformities, malignancies that arise from it can be treated through surgical procedures. The prevalence of this condition is estimated at around 1 in 100,000. It is unclear whether the men or women are more affected by this disorder due to conflicting case studies.
The disease consists of the growth multiple enchondromas which usually develop in early childhood. The growth of these enchondromas usually stops after skeletal maturation. The affected extremity is shortened (asymmetric dwarfism) and sometimes bowed due to epiphyseal fusion anomalies. Bone lesions generally present as cellular during childhood and become more solitary over time. People with Ollier disease are prone to breaking bones (fractures) and normally have swollen, aching limbs. However, many cases of solitary enchondromata go unnoticed due to lack of symptoms. Enchondromas are commonly found in the phalanges, metacarpal, and metatarsal bones in patients of Ollier disease due to the affinity of enchondromas to long tubular bones such as the femur and humerus. A unilateral distribution of bone lesions is usually observed but bilateral distributions or a singular extremity can occur as well. Approximately a third of the cases show some form of physical deformities of bowing or abnormal limb lengthening.[citation needed]
Ollier disease carries a higher risk of malignancies such as central nervous system (CNS), ovarian, and adenocarcinoma. Cranial gliomas have been linked with this disorder at an increased rate and at an earlier diagnosis age. A majority of glioma cases contain IDH gene mutations thus explaining the link between the two conditions. Juvenile granulosa cell tumour has also been associated with the disease. One case study indicates that this is due a mesodermal dysplasia in the long bones resulting in ovarian cancer.
The incidence of a secondary chondrosarcoma in Ollier disease is most commonly approximated at 25–30% with some projections even as high as 50%. Chondrosarcomas are typically developed during young adulthood and mostly form as a unifocal distribution in Ollier disease patients. The most common locations of tumors are in the pelvis and shoulder girdle. While chondrosarcoma is the most common form of a secondary malignant bone neoplasm found in cases of Ollier disease, other forms such as chordomas and osteosarcomas can occur. If left untreated, these malignant transformations may lead to fatal outcomes.[citation needed]
A related and even rarer disorder named Maffucci syndrome is a very similar condition that is characterized by the presence of multiple enchondromas with hemangiomas and occasionally lymphangiomas usually near the hands and feet but not limited to the skull, ribs, and spinal bones. This disorder is also sporadic and nonhereditary and usually detected during childhood despite being a congenital condition. Maffucci syndrome has also similarly been linked to IDH1 and IDH2 mutations specifically the IDH1 R132C hotspot mutation. This hotspot mutation is presumed to be responsible for the spindle cell hemangiomas and enchondromas in cases of Maffucci syndrome. Sanger sequencing analysis concluded that exon 4 is the primary location of mutations in IDH1 and IDH2 genes are specifically responsible for hemangiomas. Maffucci syndrome carries a significantly higher risk of malignant transformations like chondrosarcomas but also much more aggressive tumors such as acute lymphocytic leukemia and gastrointestinal and ovarian malignancies.
For many years, most research has been inconclusive regarding the cause of the disease.
Recent studies have shown that most cases of Ollier disease are believed to have been caused by isocitrate dehydrogenases IDH1 and IDH2 mutations. In one study, 35 of 43 (81%) patients with Ollier disease had either a IDH1 or IDH2 mutation. Another study suggests that R132C IDH1 mutations which are particularly dominant at exon 4 of IDH genes are linked to the growth of vascular lesions. Isocitrate deyhydrogenases IDH1 and IDH2 are catalysts responsible for the conversion of isocitrate to 2-oxoglutarate. The isocitrate deyhydrogenases IDH1 and IDH2 mutations disrupt this process resulting in unregulated production of α-ketoglutarate and a reduction in chondrocyte proliferation. In many cases of cartilaginous tumors, IDH1 and IDH2 point mutations were found thus explaining why Ollier disease is associated with many different associated conditions. Based on these case studies, most evidence suggests that the abnormal lining of lesions found in Ollier disease would suggest that the condition is caused by a post-zygotic somatic mutation thus resulting in a mosaic genetic disorder.
Approximately 8–10% of cases of patients with Ollier disease have been linked to PTH1R mutations. A particular case study the mutant heterozygous PTHR1 (R150C) receptor was observed in two unrelated patients with Ollier disease. This PTHR1 (R150C) mutant causes a reduction in chondrocyte differentiation by triggering the PTHrP-dependent pathway and decreasing PTHLH receptor function by approximately 30% creating enchondromas. One of these patients with the PTHR1 (R150C) mutant was found to have inherited the mutation from his father. This provides credence to the theory that multiple genetic mutations are needed to occur in order for Ollier disease to manifest.