Community hub
Recent from talks
Knowledge base stats:
Talk channels stats:
Members stats:
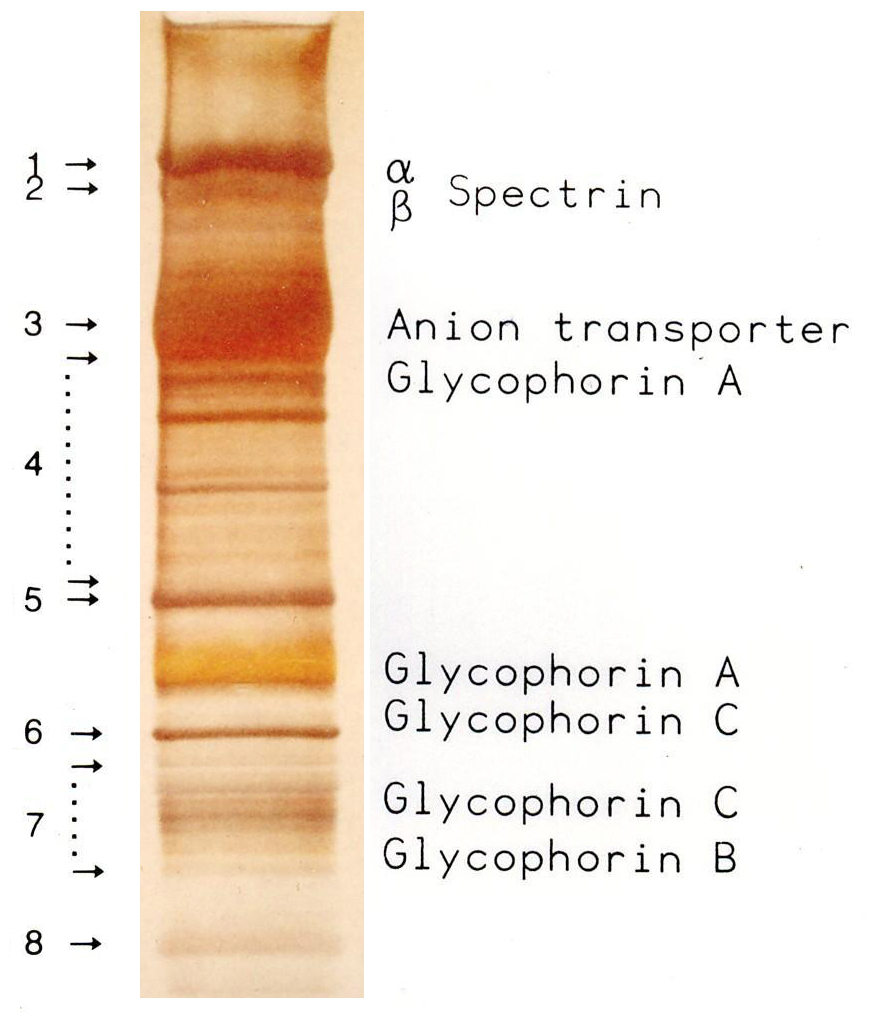
SDS-PAGE
SDS-PAGE (sodium dodecyl sulfate–polyacrylamide gel electrophoresis) is a discontinuous electrophoretic system developed by Ulrich K. Laemmli which is commonly used as a method to separate proteins with molecular masses between 5 and 250 kDa. The combined use of sodium dodecyl sulfate (SDS, also known as sodium lauryl sulfate) and polyacrylamide gel eliminates the influence of structure and charge, and proteins are separated by differences in their size. At least up to 2025, the publication describing it was the most frequently cited paper by a single author, and the second most cited overall - with over 259.000 citations.
SDS-PAGE is an electrophoresis method that allows protein separation by mass. The medium (also referred to as ′matrix′) is a polyacrylamide-based discontinuous gel. The polyacrylamide-gel is typically sandwiched between two glass plates in a slab gel. Although tube gels (in glass cylinders) were used historically, they were rapidly made obsolete with the invention of the more convenient slab gels. In addition, SDS (sodium dodecyl sulfate) is used. About 1.4 grams of SDS bind to a gram of protein, corresponding to one SDS molecule charges per two amino acids. SDS acts as a surfactant, masking the protein's intrinsic charge and conferring them very similar charge-to-mass ratios. The intrinsic charges of the proteins are negligible in comparison to the SDS loading, and the positive charges are also greatly reduced in the basic pH range of a separating gel. Upon application of a constant electric field, the proteins migrate towards the anode, each with a different speed, depending on their mass. This simple procedure allows precise protein separation by mass.
SDS tends to form spherical micelles in aqueous solutions above a certain concentration called the critical micelle concentration (CMC). Above the critical micellar concentration of 7 to 10 millimolar in solutions, the SDS simultaneously occurs as single molecules (monomer) and as micelles, below the CMC SDS occurs only as monomers in aqueous solutions. At the critical micellar concentration, a micelle consists of about 62 SDS molecules. However, only SDS monomers bind to proteins via hydrophobic interactions, whereas the SDS micelles are anionic on the outside and do not adsorb any protein. SDS is amphipathic in nature, which allows it to unfold both polar and nonpolar sections of protein structure. In SDS concentrations above 0.1 millimolar, the unfolding of proteins begins, and above 1 mM, most proteins are denatured. Due to the strong denaturing effect of SDS and the subsequent dissociation of protein complexes, quaternary structures can generally not be determined with SDS. Exceptions are proteins that are stabilised by covalent cross-linking (e.g. -S-S- linkages) and the SDS-resistant protein complexes, which are stable even in the presence of SDS (the latter, however, only at room temperature). To denature the SDS-resistant complexes a high activation energy is required, which is achieved by heating. SDS resistance is based on a metastability of the protein fold. Although the native, fully folded, SDS-resistant protein does not have sufficient stability in the presence of SDS, the chemical equilibrium of denaturation at room temperature occurs slowly. Stable protein complexes are characterised not only by SDS resistance but also by stability against proteases and an increased biological half-life.
Alternatively, polyacrylamide gel electrophoresis can also be performed with the cationic surfactants CTAB in a CTAB-PAGE, or 16-BAC in a BAC-PAGE.
The SDS-PAGE method is composed of gel preparation, sample preparation, electrophoresis, protein staining or western blotting and analysis of the generated banding pattern.
When using different buffers in the gel (discontinuous gel electrophoresis), the gels are made up to one day prior to electrophoresis, so that the diffusion does not lead to a mixing of the buffers. The gel is produced by free radical polymerization in a mold consisting of two sealed glass plates with spacers between the glass plates. In a typical mini-gel setting, the spacers have a thickness of 0.75 mm or 1.5 mm, which determines the loading capacity of the gel. For pouring the gel solution, the plates are usually clamped in a stand which temporarily seals the otherwise open underside of the glass plates with the two spacers. For the gel solution, acrylamide is mixed as gel-former (usually 4% V/V in the stacking gel and 10-12 % in the separating gel), methylenebisacrylamide as a cross-linker, stacking or separating gel buffer, water and SDS. By adding the catalyst TEMED and the radical initiator ammonium persulfate (APS) the polymerisation is started. The solution is then poured between the glass plates without creating bubbles. Depending on the amount of catalyst and radical starter and depending on the temperature, the polymerisation lasts between a quarter of an hour and several hours. The lower gel (separating gel) is poured first and covered with a few drops of a barely water-soluble alcohol (usually buffer-saturated butanol or isopropanol), which eliminates bubbles from the meniscus and protects the gel solution of the radical scavenger oxygen. After the polymerisation of the separating gel, the alcohol is discarded and the residual alcohol is removed with filter paper. After addition of APS and TEMED to the stacking gel solution, it is poured on top of the solid separation gel. Afterwards, a suitable sample comb is inserted between the glass plates without creating bubbles. The sample comb is carefully pulled out after polymerisation, leaving pockets for the sample application. For later use of proteins for protein sequencing, the gels are often prepared the day before electrophoresis to reduce reactions of unpolymerised acrylamide with cysteines in proteins.
By using a gradient mixer, gradient gels with a gradient of acrylamide (usually from 4 to 12%) can be cast, which have a larger separation range of the molecular masses. Commercial gel systems (so-called pre-cast gels) usually use the buffer substance Bis-tris methane with a pH value between 6.4 and 7.2 both in the stacking gel and in the separating gel. These gels are delivered cast and ready-to-use. Since they use only one buffer (continuous gel electrophoresis) and have a nearly neutral pH, they can be stored for several weeks. The more neutral pH slows the hydrolysis and thus the decomposition of the polyacrylamide. Furthermore, there are fewer acrylamide-modified cysteines in the proteins. Due to the constant pH in collecting and separating gel there is no stacking effect. Proteins in BisTris gels can not be stained with ruthenium complexes. This gel system has a comparatively large separation range, which can be varied by using MES or MOPS in the running buffer.
During sample preparation, the sample buffer, and thus SDS, is added in excess to the proteins, and the sample is then heated to 95 °C for five minutes, or alternatively 70 °C for ten minutes. Heating disrupts the secondary and tertiary structures of the protein by disrupting hydrogen bonds and stretching the molecules. Optionally, disulfide bridges can be cleaved by reduction. For this purpose, reducing thiols such as β-mercaptoethanol (β-ME, 5% by volume), dithiothreitol (DTT, 10–100 millimolar), dithioerythritol (DTE, 10 millimolar), tris(2-carboxyethyl)phosphine or tributylphosphine are added to the sample buffer. After cooling to room temperature, each sample is pipetted into its own well in the gel, which was previously immersed in electrophoresis buffer in the electrophoresis apparatus.
Hub AI
SDS-PAGE AI simulator
(@SDS-PAGE_simulator)
SDS-PAGE
SDS-PAGE (sodium dodecyl sulfate–polyacrylamide gel electrophoresis) is a discontinuous electrophoretic system developed by Ulrich K. Laemmli which is commonly used as a method to separate proteins with molecular masses between 5 and 250 kDa. The combined use of sodium dodecyl sulfate (SDS, also known as sodium lauryl sulfate) and polyacrylamide gel eliminates the influence of structure and charge, and proteins are separated by differences in their size. At least up to 2025, the publication describing it was the most frequently cited paper by a single author, and the second most cited overall - with over 259.000 citations.
SDS-PAGE is an electrophoresis method that allows protein separation by mass. The medium (also referred to as ′matrix′) is a polyacrylamide-based discontinuous gel. The polyacrylamide-gel is typically sandwiched between two glass plates in a slab gel. Although tube gels (in glass cylinders) were used historically, they were rapidly made obsolete with the invention of the more convenient slab gels. In addition, SDS (sodium dodecyl sulfate) is used. About 1.4 grams of SDS bind to a gram of protein, corresponding to one SDS molecule charges per two amino acids. SDS acts as a surfactant, masking the protein's intrinsic charge and conferring them very similar charge-to-mass ratios. The intrinsic charges of the proteins are negligible in comparison to the SDS loading, and the positive charges are also greatly reduced in the basic pH range of a separating gel. Upon application of a constant electric field, the proteins migrate towards the anode, each with a different speed, depending on their mass. This simple procedure allows precise protein separation by mass.
SDS tends to form spherical micelles in aqueous solutions above a certain concentration called the critical micelle concentration (CMC). Above the critical micellar concentration of 7 to 10 millimolar in solutions, the SDS simultaneously occurs as single molecules (monomer) and as micelles, below the CMC SDS occurs only as monomers in aqueous solutions. At the critical micellar concentration, a micelle consists of about 62 SDS molecules. However, only SDS monomers bind to proteins via hydrophobic interactions, whereas the SDS micelles are anionic on the outside and do not adsorb any protein. SDS is amphipathic in nature, which allows it to unfold both polar and nonpolar sections of protein structure. In SDS concentrations above 0.1 millimolar, the unfolding of proteins begins, and above 1 mM, most proteins are denatured. Due to the strong denaturing effect of SDS and the subsequent dissociation of protein complexes, quaternary structures can generally not be determined with SDS. Exceptions are proteins that are stabilised by covalent cross-linking (e.g. -S-S- linkages) and the SDS-resistant protein complexes, which are stable even in the presence of SDS (the latter, however, only at room temperature). To denature the SDS-resistant complexes a high activation energy is required, which is achieved by heating. SDS resistance is based on a metastability of the protein fold. Although the native, fully folded, SDS-resistant protein does not have sufficient stability in the presence of SDS, the chemical equilibrium of denaturation at room temperature occurs slowly. Stable protein complexes are characterised not only by SDS resistance but also by stability against proteases and an increased biological half-life.
Alternatively, polyacrylamide gel electrophoresis can also be performed with the cationic surfactants CTAB in a CTAB-PAGE, or 16-BAC in a BAC-PAGE.
The SDS-PAGE method is composed of gel preparation, sample preparation, electrophoresis, protein staining or western blotting and analysis of the generated banding pattern.
When using different buffers in the gel (discontinuous gel electrophoresis), the gels are made up to one day prior to electrophoresis, so that the diffusion does not lead to a mixing of the buffers. The gel is produced by free radical polymerization in a mold consisting of two sealed glass plates with spacers between the glass plates. In a typical mini-gel setting, the spacers have a thickness of 0.75 mm or 1.5 mm, which determines the loading capacity of the gel. For pouring the gel solution, the plates are usually clamped in a stand which temporarily seals the otherwise open underside of the glass plates with the two spacers. For the gel solution, acrylamide is mixed as gel-former (usually 4% V/V in the stacking gel and 10-12 % in the separating gel), methylenebisacrylamide as a cross-linker, stacking or separating gel buffer, water and SDS. By adding the catalyst TEMED and the radical initiator ammonium persulfate (APS) the polymerisation is started. The solution is then poured between the glass plates without creating bubbles. Depending on the amount of catalyst and radical starter and depending on the temperature, the polymerisation lasts between a quarter of an hour and several hours. The lower gel (separating gel) is poured first and covered with a few drops of a barely water-soluble alcohol (usually buffer-saturated butanol or isopropanol), which eliminates bubbles from the meniscus and protects the gel solution of the radical scavenger oxygen. After the polymerisation of the separating gel, the alcohol is discarded and the residual alcohol is removed with filter paper. After addition of APS and TEMED to the stacking gel solution, it is poured on top of the solid separation gel. Afterwards, a suitable sample comb is inserted between the glass plates without creating bubbles. The sample comb is carefully pulled out after polymerisation, leaving pockets for the sample application. For later use of proteins for protein sequencing, the gels are often prepared the day before electrophoresis to reduce reactions of unpolymerised acrylamide with cysteines in proteins.
By using a gradient mixer, gradient gels with a gradient of acrylamide (usually from 4 to 12%) can be cast, which have a larger separation range of the molecular masses. Commercial gel systems (so-called pre-cast gels) usually use the buffer substance Bis-tris methane with a pH value between 6.4 and 7.2 both in the stacking gel and in the separating gel. These gels are delivered cast and ready-to-use. Since they use only one buffer (continuous gel electrophoresis) and have a nearly neutral pH, they can be stored for several weeks. The more neutral pH slows the hydrolysis and thus the decomposition of the polyacrylamide. Furthermore, there are fewer acrylamide-modified cysteines in the proteins. Due to the constant pH in collecting and separating gel there is no stacking effect. Proteins in BisTris gels can not be stained with ruthenium complexes. This gel system has a comparatively large separation range, which can be varied by using MES or MOPS in the running buffer.
During sample preparation, the sample buffer, and thus SDS, is added in excess to the proteins, and the sample is then heated to 95 °C for five minutes, or alternatively 70 °C for ten minutes. Heating disrupts the secondary and tertiary structures of the protein by disrupting hydrogen bonds and stretching the molecules. Optionally, disulfide bridges can be cleaved by reduction. For this purpose, reducing thiols such as β-mercaptoethanol (β-ME, 5% by volume), dithiothreitol (DTT, 10–100 millimolar), dithioerythritol (DTE, 10 millimolar), tris(2-carboxyethyl)phosphine or tributylphosphine are added to the sample buffer. After cooling to room temperature, each sample is pipetted into its own well in the gel, which was previously immersed in electrophoresis buffer in the electrophoresis apparatus.