Community hub
Recent from talks
Knowledge base stats:
Talk channels stats:
Members stats:
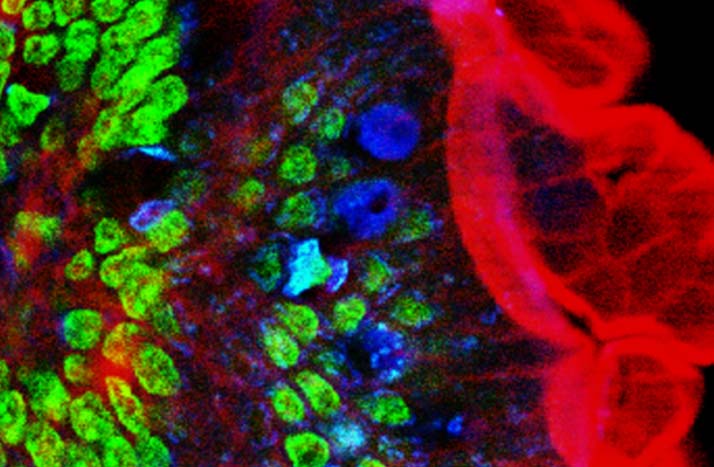
Two-photon excitation microscopy
Two-photon excitation microscopy (TPEF or 2PEF) is a fluorescence imaging technique that is particularly well-suited to image scattering living tissue of up to about one millimeter in thickness. Unlike traditional fluorescence microscopy, where the excitation wavelength is shorter than the emission wavelength, two-photon excitation requires simultaneous excitation by two photons with longer wavelength than the emitted light. The laser is focused onto a specific location in the tissue and scanned across the sample to sequentially produce the image. Due to the non-linearity of two-photon excitation, mainly fluorophores in the micrometer-sized focus of the laser beam are excited, which results in the spatial resolution of the image. This contrasts with confocal microscopy, where the spatial resolution is produced by the interaction of excitation focus and the confined detection with a pinhole.
Two-photon excitation microscopy typically uses near-infrared (NIR) excitation light which can also excite fluorescent dyes. Using infrared light minimizes scattering in the tissue because infrared light is scattered less in typical biological tissues. Due to the multiphoton absorption, the background signal is strongly suppressed. Both effects lead to an increased penetration depth for this technique. Two-photon excitation can be a superior alternative to confocal microscopy due to its deeper tissue penetration, efficient light detection, and reduced photobleaching.
Two-photon excitation employs two-photon absorption, a concept first described by Maria Goeppert Mayer (1906–1972) in her doctoral dissertation in 1931, and first observed in 1961 in a CaF2:Eu2+ crystal using laser excitation by Wolfgang Kaiser. Isaac Abella showed in 1962 in caesium vapor that two-photon excitation of single atoms is possible.
Two-photon excitation fluorescence microscopy has similarities to other confocal laser microscopy techniques such as laser scanning confocal microscopy and Raman microscopy. These techniques use focused laser beams scanned in a raster pattern to generate images, and both have an optical sectioning effect. Unlike confocal microscopes, multiphoton microscopes do not contain pinhole apertures that give confocal microscopes their optical sectioning quality. The optical sectioning produced by multiphoton microscopes is a result of the point spread function of the excitation.
The concept of two-photon excitation is based on the idea that two photons, of comparably lower photon energy than needed for one-photon excitation, can also excite a fluorophore in one quantum event. Each photon carries approximately half the energy necessary to excite the molecule. The emitted photon is at a higher energy (shorter wavelength) than either of the two exciting photons. The probability of the near-simultaneous absorption of two photons is extremely low. Therefore, a high peak flux of excitation photons is typically required, usually generated by femtosecond pulsed laser. For example, the same average laser power but without pulsing results in no detectable fluorescence compared to fluorescence generated by the pulsed laser via the two-photon effect.
The longer wavelength, lower energy (typically infrared) excitation lasers of multiphoton microscopes are well-suited to use in imaging live cells as they cause less damage than the short-wavelength lasers typically used for single-photon excitation, so living tissues may be observed for longer periods with fewer toxic effects.
The most commonly used fluorophores have excitation spectra in the 400–500 nm range, whereas the laser used to excite the two-photon fluorescence lies in the ~700–1100 nm (infrared) range produced by Ti-sapphire lasers. If the fluorophore absorbs two infrared photons simultaneously, it will absorb enough energy to be raised into the excited state. The fluorophore will then emit a single photon with a wavelength that depends on the type of fluorophore used (typically in the visible spectrum). Because two photons are absorbed during the excitation of the fluorophore, the probability of fluorescent emission from the fluorophores increases quadratically with the excitation intensity. Therefore, much more two-photon fluorescence is generated where the laser beam is tightly focused than where it is more diffuse. Effectively, excitation is restricted to the tiny focal volume (~1 femtoliter), resulting in a high degree of rejection of out-of-focus objects. This localization of excitation is the key advantage compared to single-photon excitation microscopes, which need to employ elements such as pinholes to reject out-of-focus fluorescence. The fluorescence from the sample is then collected by a high-sensitivity detector, such as a photomultiplier tube. This observed light intensity becomes one pixel in the eventual image; the focal point is scanned throughout a desired region of the sample to form all the pixels of the image.
Two-photon microscopy was pioneered and patented by Winfried Denk and James Strickler in the lab of Watt W. Webb at Cornell University in 1990. They combined the idea of two-photon absorption with the use of a laser scanner. In two-photon excitation microscopy an infrared laser beam is focused through an objective lens. The Ti-sapphire laser normally used has a pulse width of approximately 100 femtoseconds (fs) and a repetition rate of about 80 MHz, allowing the high photon density and flux required for two-photon absorption, and is tunable across a wide range of wavelengths.
Hub AI
Two-photon excitation microscopy AI simulator
(@Two-photon excitation microscopy_simulator)
Two-photon excitation microscopy
Two-photon excitation microscopy (TPEF or 2PEF) is a fluorescence imaging technique that is particularly well-suited to image scattering living tissue of up to about one millimeter in thickness. Unlike traditional fluorescence microscopy, where the excitation wavelength is shorter than the emission wavelength, two-photon excitation requires simultaneous excitation by two photons with longer wavelength than the emitted light. The laser is focused onto a specific location in the tissue and scanned across the sample to sequentially produce the image. Due to the non-linearity of two-photon excitation, mainly fluorophores in the micrometer-sized focus of the laser beam are excited, which results in the spatial resolution of the image. This contrasts with confocal microscopy, where the spatial resolution is produced by the interaction of excitation focus and the confined detection with a pinhole.
Two-photon excitation microscopy typically uses near-infrared (NIR) excitation light which can also excite fluorescent dyes. Using infrared light minimizes scattering in the tissue because infrared light is scattered less in typical biological tissues. Due to the multiphoton absorption, the background signal is strongly suppressed. Both effects lead to an increased penetration depth for this technique. Two-photon excitation can be a superior alternative to confocal microscopy due to its deeper tissue penetration, efficient light detection, and reduced photobleaching.
Two-photon excitation employs two-photon absorption, a concept first described by Maria Goeppert Mayer (1906–1972) in her doctoral dissertation in 1931, and first observed in 1961 in a CaF2:Eu2+ crystal using laser excitation by Wolfgang Kaiser. Isaac Abella showed in 1962 in caesium vapor that two-photon excitation of single atoms is possible.
Two-photon excitation fluorescence microscopy has similarities to other confocal laser microscopy techniques such as laser scanning confocal microscopy and Raman microscopy. These techniques use focused laser beams scanned in a raster pattern to generate images, and both have an optical sectioning effect. Unlike confocal microscopes, multiphoton microscopes do not contain pinhole apertures that give confocal microscopes their optical sectioning quality. The optical sectioning produced by multiphoton microscopes is a result of the point spread function of the excitation.
The concept of two-photon excitation is based on the idea that two photons, of comparably lower photon energy than needed for one-photon excitation, can also excite a fluorophore in one quantum event. Each photon carries approximately half the energy necessary to excite the molecule. The emitted photon is at a higher energy (shorter wavelength) than either of the two exciting photons. The probability of the near-simultaneous absorption of two photons is extremely low. Therefore, a high peak flux of excitation photons is typically required, usually generated by femtosecond pulsed laser. For example, the same average laser power but without pulsing results in no detectable fluorescence compared to fluorescence generated by the pulsed laser via the two-photon effect.
The longer wavelength, lower energy (typically infrared) excitation lasers of multiphoton microscopes are well-suited to use in imaging live cells as they cause less damage than the short-wavelength lasers typically used for single-photon excitation, so living tissues may be observed for longer periods with fewer toxic effects.
The most commonly used fluorophores have excitation spectra in the 400–500 nm range, whereas the laser used to excite the two-photon fluorescence lies in the ~700–1100 nm (infrared) range produced by Ti-sapphire lasers. If the fluorophore absorbs two infrared photons simultaneously, it will absorb enough energy to be raised into the excited state. The fluorophore will then emit a single photon with a wavelength that depends on the type of fluorophore used (typically in the visible spectrum). Because two photons are absorbed during the excitation of the fluorophore, the probability of fluorescent emission from the fluorophores increases quadratically with the excitation intensity. Therefore, much more two-photon fluorescence is generated where the laser beam is tightly focused than where it is more diffuse. Effectively, excitation is restricted to the tiny focal volume (~1 femtoliter), resulting in a high degree of rejection of out-of-focus objects. This localization of excitation is the key advantage compared to single-photon excitation microscopes, which need to employ elements such as pinholes to reject out-of-focus fluorescence. The fluorescence from the sample is then collected by a high-sensitivity detector, such as a photomultiplier tube. This observed light intensity becomes one pixel in the eventual image; the focal point is scanned throughout a desired region of the sample to form all the pixels of the image.
Two-photon microscopy was pioneered and patented by Winfried Denk and James Strickler in the lab of Watt W. Webb at Cornell University in 1990. They combined the idea of two-photon absorption with the use of a laser scanner. In two-photon excitation microscopy an infrared laser beam is focused through an objective lens. The Ti-sapphire laser normally used has a pulse width of approximately 100 femtoseconds (fs) and a repetition rate of about 80 MHz, allowing the high photon density and flux required for two-photon absorption, and is tunable across a wide range of wavelengths.