
Community hub
Recent from talks
Contribute something to knowledge base
Content stats: 0 posts, 0 articles, 1 media, 0 notes
Members stats: 0 subscribers, 0 contributors, 0 moderators, 0 supporters
Subscribers
Supporters
Contributors
Moderators
Hub AI
CYP2D6 AI simulator
(@CYP2D6_simulator)
Hub AI
CYP2D6 AI simulator
(@CYP2D6_simulator)
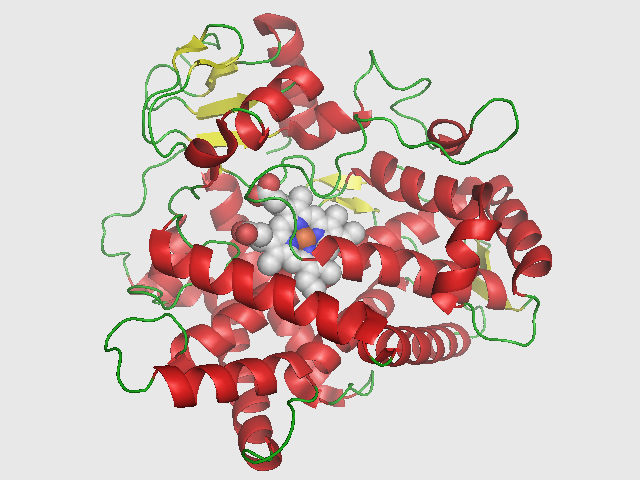
CYP2D6
Cytochrome P450 2D6 (CYP2D6) is an enzyme that in humans is encoded by the CYP2D6 gene. CYP2D6 is primarily expressed in the liver. It is also highly expressed in areas of the central nervous system, including the substantia nigra.
CYP2D6, a member of the cytochrome P450 mixed-function oxidase system, is one of the most important enzymes involved in the metabolism of xenobiotics in the body. In particular, CYP2D6 is responsible for the metabolism and elimination of approximately 25% of clinically used drugs, via the addition or removal of certain functional groups – specifically, hydroxylation, demethylation, and dealkylation. CYP2D6 also activates some prodrugs. This enzyme also metabolizes several endogenous substances, such as N,N-Dimethyltryptamine, hydroxytryptamines, neurosteroids, and both m-tyramine and p-tyramine which CYP2D6 metabolizes into dopamine in the brain and liver.
Considerable variation exists in the efficiency and amount of CYP2D6 enzyme produced between individuals. Hence, for drugs that are metabolized by CYP2D6 (that is, drugs that are CYP2D6 substrates), certain individuals will eliminate these drugs quickly (ultrarapid metabolizers) while others slowly (poor metabolizers). If a drug is metabolized quickly, the drug's efficacy may decrease, while if a drug is metabolized too slowly, toxicity may result. The dose of the drug may have to be adjusted to take into account of the speed at which it is metabolized by CYP2D6. People who more rapidly metabolize prodrugs, such as codeine or tramadol, reach higher-than-therapeutic levels. A case study of the death of an infant breastfed by an ultrarapid metabolizer mother taking codeine impacted postnatal pain relief clinical practices, but was later debunked. These drugs may also cause serious toxicity in ultrarapid metabolizer patients when used to treat other post-operative pain, such as after tonsillectomy. Other drugs may function as inhibitors of CYP2D6 activity or inducers of CYP2D6 enzyme expression that will lead to decreased or increased CYP2D6 activity respectively. If such a drug is taken at the same time as a second drug that is a CYP2D6 substrate, the first drug may affect the elimination rate of the second through what is known as a drug-drug interaction.
The gene is located on chromosome 22q13.1. near two cytochrome P450 pseudogenes (CYP2D7P and CYP2D8P). Among them, CYP2D7P originated from CYP2D6 in a stem lineage of great apes and humans, the CYP2D8P originated from CYP2D6 in a stem lineage of Catarrhine and New World monkeys' stem lineage. Alternatively spliced transcript variants encoding different isoforms have been found for this gene.
CYP2D6 shows the largest phenotypical variability among the CYPs, largely due to genetic polymorphism. The genotype accounts for normal, reduced, and non-existent CYP2D6 function in subjects. Pharmacogenomic tests are now available to identify patients with variations in the CYP2D6 allele and have been shown to have widespread use in clinical practice. The CYP2D6 function in any particular subject may be described as one of the following:
A patient's CYP2D6 phenotype is often clinically determined via the administration of debrisoquine (a selective CYP2D6 substrate) and subsequent plasma concentration assay of the debrisoquine metabolite (4-hydroxydebrisoquine).
The type of CYP2D6 function of an individual may influence the person's response to different doses of drugs that CYP2D6 metabolizes. The nature of the effect on the drug response depends not only on the type of CYP2D6 function, but also on the extent to which processing of the drug by CYP2D6 results in a chemical that has an effect that is similar, stronger, or weaker than the original drug, or no effect at all. For example, if CYP2D6 converts a drug that has a strong effect into a substance that has a weaker effect, then poor metabolizers (weak CYP2D6 function) will have an exaggerated response to the drug and stronger side-effects; conversely, if CYP2D6 converts a different drug into a substance that has a greater effect than its parent chemical, then ultrarapid metabolizers (strong CYP2D6 function) will have an exaggerated response to the drug and stronger side-effects. Information about how human genetic variation of CYP2D6 affects response to medications can be found in databases such PharmGKB, Clinical Pharmacogenetics Implementation Consortium (CPIC).
The variability in metabolism is due to multiple different polymorphisms of the CYP2D6 allele, located on chromosome 22. Subjects possessing certain allelic variants will show normal, decreased, or no CYP2D6 function, depending on the allele. Pharmacogenomic tests are now available to identify patients with variations in the CYP2D6 allele and have been shown to have widespread use in clinical practice. The current known alleles of CYP2D6 and their clinical function can be found in databases such as PharmVar.
CYP2D6
Cytochrome P450 2D6 (CYP2D6) is an enzyme that in humans is encoded by the CYP2D6 gene. CYP2D6 is primarily expressed in the liver. It is also highly expressed in areas of the central nervous system, including the substantia nigra.
CYP2D6, a member of the cytochrome P450 mixed-function oxidase system, is one of the most important enzymes involved in the metabolism of xenobiotics in the body. In particular, CYP2D6 is responsible for the metabolism and elimination of approximately 25% of clinically used drugs, via the addition or removal of certain functional groups – specifically, hydroxylation, demethylation, and dealkylation. CYP2D6 also activates some prodrugs. This enzyme also metabolizes several endogenous substances, such as N,N-Dimethyltryptamine, hydroxytryptamines, neurosteroids, and both m-tyramine and p-tyramine which CYP2D6 metabolizes into dopamine in the brain and liver.
Considerable variation exists in the efficiency and amount of CYP2D6 enzyme produced between individuals. Hence, for drugs that are metabolized by CYP2D6 (that is, drugs that are CYP2D6 substrates), certain individuals will eliminate these drugs quickly (ultrarapid metabolizers) while others slowly (poor metabolizers). If a drug is metabolized quickly, the drug's efficacy may decrease, while if a drug is metabolized too slowly, toxicity may result. The dose of the drug may have to be adjusted to take into account of the speed at which it is metabolized by CYP2D6. People who more rapidly metabolize prodrugs, such as codeine or tramadol, reach higher-than-therapeutic levels. A case study of the death of an infant breastfed by an ultrarapid metabolizer mother taking codeine impacted postnatal pain relief clinical practices, but was later debunked. These drugs may also cause serious toxicity in ultrarapid metabolizer patients when used to treat other post-operative pain, such as after tonsillectomy. Other drugs may function as inhibitors of CYP2D6 activity or inducers of CYP2D6 enzyme expression that will lead to decreased or increased CYP2D6 activity respectively. If such a drug is taken at the same time as a second drug that is a CYP2D6 substrate, the first drug may affect the elimination rate of the second through what is known as a drug-drug interaction.
The gene is located on chromosome 22q13.1. near two cytochrome P450 pseudogenes (CYP2D7P and CYP2D8P). Among them, CYP2D7P originated from CYP2D6 in a stem lineage of great apes and humans, the CYP2D8P originated from CYP2D6 in a stem lineage of Catarrhine and New World monkeys' stem lineage. Alternatively spliced transcript variants encoding different isoforms have been found for this gene.
CYP2D6 shows the largest phenotypical variability among the CYPs, largely due to genetic polymorphism. The genotype accounts for normal, reduced, and non-existent CYP2D6 function in subjects. Pharmacogenomic tests are now available to identify patients with variations in the CYP2D6 allele and have been shown to have widespread use in clinical practice. The CYP2D6 function in any particular subject may be described as one of the following:
A patient's CYP2D6 phenotype is often clinically determined via the administration of debrisoquine (a selective CYP2D6 substrate) and subsequent plasma concentration assay of the debrisoquine metabolite (4-hydroxydebrisoquine).
The type of CYP2D6 function of an individual may influence the person's response to different doses of drugs that CYP2D6 metabolizes. The nature of the effect on the drug response depends not only on the type of CYP2D6 function, but also on the extent to which processing of the drug by CYP2D6 results in a chemical that has an effect that is similar, stronger, or weaker than the original drug, or no effect at all. For example, if CYP2D6 converts a drug that has a strong effect into a substance that has a weaker effect, then poor metabolizers (weak CYP2D6 function) will have an exaggerated response to the drug and stronger side-effects; conversely, if CYP2D6 converts a different drug into a substance that has a greater effect than its parent chemical, then ultrarapid metabolizers (strong CYP2D6 function) will have an exaggerated response to the drug and stronger side-effects. Information about how human genetic variation of CYP2D6 affects response to medications can be found in databases such PharmGKB, Clinical Pharmacogenetics Implementation Consortium (CPIC).
The variability in metabolism is due to multiple different polymorphisms of the CYP2D6 allele, located on chromosome 22. Subjects possessing certain allelic variants will show normal, decreased, or no CYP2D6 function, depending on the allele. Pharmacogenomic tests are now available to identify patients with variations in the CYP2D6 allele and have been shown to have widespread use in clinical practice. The current known alleles of CYP2D6 and their clinical function can be found in databases such as PharmVar.
Recent media
Recent media