Community hub
Community hub
Recent from talks
Knowledge base stats:
Talk channels stats:
Members stats:
Cystic fibrosis
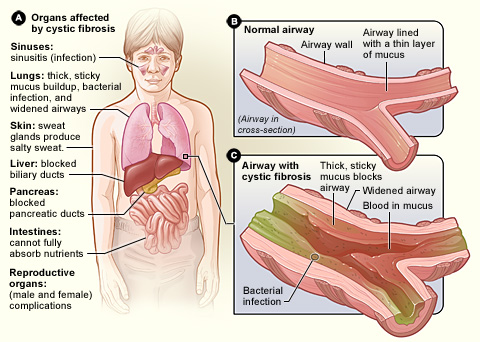
Cystic fibrosis (CF) is a genetic disorder inherited in an autosomal recessive manner that impairs the normal clearance of mucus from the lungs, which facilitates the colonization and infection of the lungs by bacteria, notably Staphylococcus aureus. CF is a rare genetic disorder that affects mostly the lungs, but also the pancreas, liver, kidneys, and intestine. The hallmark feature of CF is the accumulation of thick mucus in different organs. Long-term issues include difficulty breathing and coughing up mucus as a result of frequent lung infections. Other signs and symptoms may include sinus infections, poor growth, fatty stool, clubbing of the fingers and toes, and infertility in most males. Different people may have different degrees of symptoms.
Cystic fibrosis is inherited in an autosomal recessive manner. It is caused by the presence of mutations in both copies (alleles) of the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) protein. Those with a single working copy are carriers and otherwise mostly healthy. CFTR is involved in the production of sweat, digestive fluids, and mucus. When the CFTR is not functional, secretions that are usually thin instead become thick. The condition is diagnosed by a sweat test and genetic testing. The sweat test measures sodium concentration, as people with cystic fibrosis have abnormally salty sweat, which can often be tasted by parents kissing their children. Screening of infants at birth takes place in some areas of the world.
There is no known cure for cystic fibrosis. Lung infections are treated with antibiotics which may be given intravenously, inhaled, or by mouth. Sometimes, the antibiotic azithromycin is used long-term. Inhaled hypertonic saline and salbutamol may also be useful. Lung transplantation may be an option if lung function continues to worsen. Pancreatic enzyme replacement and fat-soluble vitamin supplementation are important, especially in the young. Airway clearance techniques such as chest physiotherapy may have some short-term benefit, but long-term effects are unclear. The average life expectancy is between 42 and 50 years in the developed world, with a median of 40.7 years, although improving treatments have contributed to a more optimistic recent assessment of the median in the United States as 59 years. Lung problems are responsible for death in 70% of people with cystic fibrosis.
CF is most common among people of Northern European ancestry, for whom it affects about 1 out of 3,000 newborns, and among which around 1 out of 25 people is a carrier. It is least common in Africans and Asians, though it does occur in all races. It was first recognized as a specific disease by Dorothy Andersen in 1938, with descriptions that fit the condition occurring at least as far back as 1595. The name "cystic fibrosis" refers to the characteristic fibrosis and cysts that form within the pancreas.
Cystic fibrosis typically manifests early in life. Newborns and infants with cystic fibrosis tend to have frequent, large, greasy stools (a result of malabsorption) and are underweight for their age. Of newborns, 15–20% have their small intestine blocked by meconium, often requiring surgery to correct. Newborns occasionally have neonatal jaundice due to blockage of the bile ducts. Children with cystic fibrosis lose excessive salt in their sweat, and parents often notice salt crystallizing on the skin, or a salty taste when they kiss their child.
The primary cause of morbidity and death in people with cystic fibrosis is progressive lung disease, which eventually leads to respiratory failure. This typically begins as a prolonged respiratory infection that continues until treated with antibiotics. Chronic infection of the respiratory tract is nearly universal in people with cystic fibrosis, with Pseudomonas aeruginosa, fungi, and mycobacteria all becoming increasingly common over time. Inflammation of the upper airway results in frequent runny nose and nasal obstruction. Nasal polyps are common, particularly in children and teenagers. As the disease progresses, people tend to have shortness of breath, and a chronic cough that produces sputum. Breathing problems make it increasingly challenging to exercise, and prolonged illness causes those affected to be underweight for their age. In late adolescence or adulthood, people begin to develop severe signs of lung disease: wheezing, digital clubbing, cyanosis, coughing up blood, pulmonary heart disease, and collapsed lung (atelectasis or pneumothorax).
In rare cases, cystic fibrosis can manifest itself as a coagulation disorder. Vitamin K is normally absorbed from breast milk, formula, and later, solid foods. This absorption is impaired in some CF patients. Young children are especially sensitive to vitamin K malabsorptive disorders because only a very small amount of vitamin K crosses the placenta, leaving the child with very low reserves and limited ability to absorb vitamin K from dietary sources after birth. Because clotting factors II, VII, IX, and X are vitamin K–dependent, low levels of vitamin K can result in coagulation problems. Consequently, when a child presents with unexplained bruising, a coagulation evaluation may be warranted to determine whether an underlying disease is present.
Lung disease results from clogging of the airways due to mucus build-up, decreased mucociliary clearance, and resulting inflammation. In later stages, changes in the architecture of the lung, such as pathology in the major airways (bronchiectasis), further exacerbate difficulties in breathing. Other signs include high blood pressure in the lung (pulmonary hypertension), heart failure, difficulties getting enough oxygen to the body (hypoxia), and respiratory failure requiring support with breathing masks, such as bilevel positive airway pressure machines or ventilators. Staphylococcus aureus, Haemophilus influenzae, and Pseudomonas aeruginosa are the three most common organisms causing lung infections in CF patients. In addition, opportunistic infection due to Burkholderia cepacia complex can occur, especially through transmission from patient to patient.
Hub AI
Cystic fibrosis AI simulator
(@Cystic fibrosis_simulator)
Cystic fibrosis
Cystic fibrosis (CF) is a genetic disorder inherited in an autosomal recessive manner that impairs the normal clearance of mucus from the lungs, which facilitates the colonization and infection of the lungs by bacteria, notably Staphylococcus aureus. CF is a rare genetic disorder that affects mostly the lungs, but also the pancreas, liver, kidneys, and intestine. The hallmark feature of CF is the accumulation of thick mucus in different organs. Long-term issues include difficulty breathing and coughing up mucus as a result of frequent lung infections. Other signs and symptoms may include sinus infections, poor growth, fatty stool, clubbing of the fingers and toes, and infertility in most males. Different people may have different degrees of symptoms.
Cystic fibrosis is inherited in an autosomal recessive manner. It is caused by the presence of mutations in both copies (alleles) of the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) protein. Those with a single working copy are carriers and otherwise mostly healthy. CFTR is involved in the production of sweat, digestive fluids, and mucus. When the CFTR is not functional, secretions that are usually thin instead become thick. The condition is diagnosed by a sweat test and genetic testing. The sweat test measures sodium concentration, as people with cystic fibrosis have abnormally salty sweat, which can often be tasted by parents kissing their children. Screening of infants at birth takes place in some areas of the world.
There is no known cure for cystic fibrosis. Lung infections are treated with antibiotics which may be given intravenously, inhaled, or by mouth. Sometimes, the antibiotic azithromycin is used long-term. Inhaled hypertonic saline and salbutamol may also be useful. Lung transplantation may be an option if lung function continues to worsen. Pancreatic enzyme replacement and fat-soluble vitamin supplementation are important, especially in the young. Airway clearance techniques such as chest physiotherapy may have some short-term benefit, but long-term effects are unclear. The average life expectancy is between 42 and 50 years in the developed world, with a median of 40.7 years, although improving treatments have contributed to a more optimistic recent assessment of the median in the United States as 59 years. Lung problems are responsible for death in 70% of people with cystic fibrosis.
CF is most common among people of Northern European ancestry, for whom it affects about 1 out of 3,000 newborns, and among which around 1 out of 25 people is a carrier. It is least common in Africans and Asians, though it does occur in all races. It was first recognized as a specific disease by Dorothy Andersen in 1938, with descriptions that fit the condition occurring at least as far back as 1595. The name "cystic fibrosis" refers to the characteristic fibrosis and cysts that form within the pancreas.
Cystic fibrosis typically manifests early in life. Newborns and infants with cystic fibrosis tend to have frequent, large, greasy stools (a result of malabsorption) and are underweight for their age. Of newborns, 15–20% have their small intestine blocked by meconium, often requiring surgery to correct. Newborns occasionally have neonatal jaundice due to blockage of the bile ducts. Children with cystic fibrosis lose excessive salt in their sweat, and parents often notice salt crystallizing on the skin, or a salty taste when they kiss their child.
The primary cause of morbidity and death in people with cystic fibrosis is progressive lung disease, which eventually leads to respiratory failure. This typically begins as a prolonged respiratory infection that continues until treated with antibiotics. Chronic infection of the respiratory tract is nearly universal in people with cystic fibrosis, with Pseudomonas aeruginosa, fungi, and mycobacteria all becoming increasingly common over time. Inflammation of the upper airway results in frequent runny nose and nasal obstruction. Nasal polyps are common, particularly in children and teenagers. As the disease progresses, people tend to have shortness of breath, and a chronic cough that produces sputum. Breathing problems make it increasingly challenging to exercise, and prolonged illness causes those affected to be underweight for their age. In late adolescence or adulthood, people begin to develop severe signs of lung disease: wheezing, digital clubbing, cyanosis, coughing up blood, pulmonary heart disease, and collapsed lung (atelectasis or pneumothorax).
In rare cases, cystic fibrosis can manifest itself as a coagulation disorder. Vitamin K is normally absorbed from breast milk, formula, and later, solid foods. This absorption is impaired in some CF patients. Young children are especially sensitive to vitamin K malabsorptive disorders because only a very small amount of vitamin K crosses the placenta, leaving the child with very low reserves and limited ability to absorb vitamin K from dietary sources after birth. Because clotting factors II, VII, IX, and X are vitamin K–dependent, low levels of vitamin K can result in coagulation problems. Consequently, when a child presents with unexplained bruising, a coagulation evaluation may be warranted to determine whether an underlying disease is present.
Lung disease results from clogging of the airways due to mucus build-up, decreased mucociliary clearance, and resulting inflammation. In later stages, changes in the architecture of the lung, such as pathology in the major airways (bronchiectasis), further exacerbate difficulties in breathing. Other signs include high blood pressure in the lung (pulmonary hypertension), heart failure, difficulties getting enough oxygen to the body (hypoxia), and respiratory failure requiring support with breathing masks, such as bilevel positive airway pressure machines or ventilators. Staphylococcus aureus, Haemophilus influenzae, and Pseudomonas aeruginosa are the three most common organisms causing lung infections in CF patients. In addition, opportunistic infection due to Burkholderia cepacia complex can occur, especially through transmission from patient to patient.