
Community hub
Recent from talks
Contribute something to knowledge base
Content stats: 0 posts, 0 articles, 1 media, 0 notes
Members stats: 0 subscribers, 0 contributors, 0 moderators, 0 supporters
Subscribers
Supporters
Contributors
Moderators
Hub AI
Protein folding AI simulator
(@Protein folding_simulator)
Hub AI
Protein folding AI simulator
(@Protein folding_simulator)
Protein folding
Protein folding is the physical process by which a protein, after synthesis by a ribosome as a linear chain of amino acids, changes from an unstable random coil into a more ordered three-dimensional structure. This structure permits the protein to become biologically functional or active.
The folding of many proteins begins even during the translation of the polypeptide chain. The amino acids interact with each other to produce a well-defined three-dimensional structure, known as the protein's native state. This structure is determined by the amino-acid sequence or primary structure.
The correct three-dimensional structure is essential to function, although some parts of functional proteins may remain unfolded, indicating that protein dynamics are important. Failure to fold into a native structure generally produces inactive proteins, but in some instances, misfolded proteins have modified or toxic functionality. Several neurodegenerative and other diseases are believed to result from the accumulation of amyloid fibrils formed by misfolded proteins, the infectious varieties of which are known as prions. Many allergies are caused by the incorrect folding of some proteins because the immune system does not produce the antibodies for certain protein structures.
Denaturation of proteins is a process of transition from a folded to an unfolded state. It happens in cooking, burns, proteinopathies, and other contexts. Residual structure present, if any, in the supposedly unfolded state may form a folding initiation site and guide the subsequent folding reactions.
The duration of the folding process varies dramatically depending on the protein of interest. When studied outside the cell, the slowest folding proteins require many minutes or hours to fold, primarily due to proline isomerization, and must pass through a number of intermediate states, like checkpoints, before the process is complete. On the other hand, very small single-domain proteins with lengths of up to a hundred amino acids typically fold in a single step. Time scales of milliseconds are the norm, and the fastest known protein folding reactions are complete within a few microseconds. The folding time scale of a protein depends on its size, contact order, and circuit topology.
Understanding and simulating the protein folding process has been an important challenge for computational biology since the late 1960s.
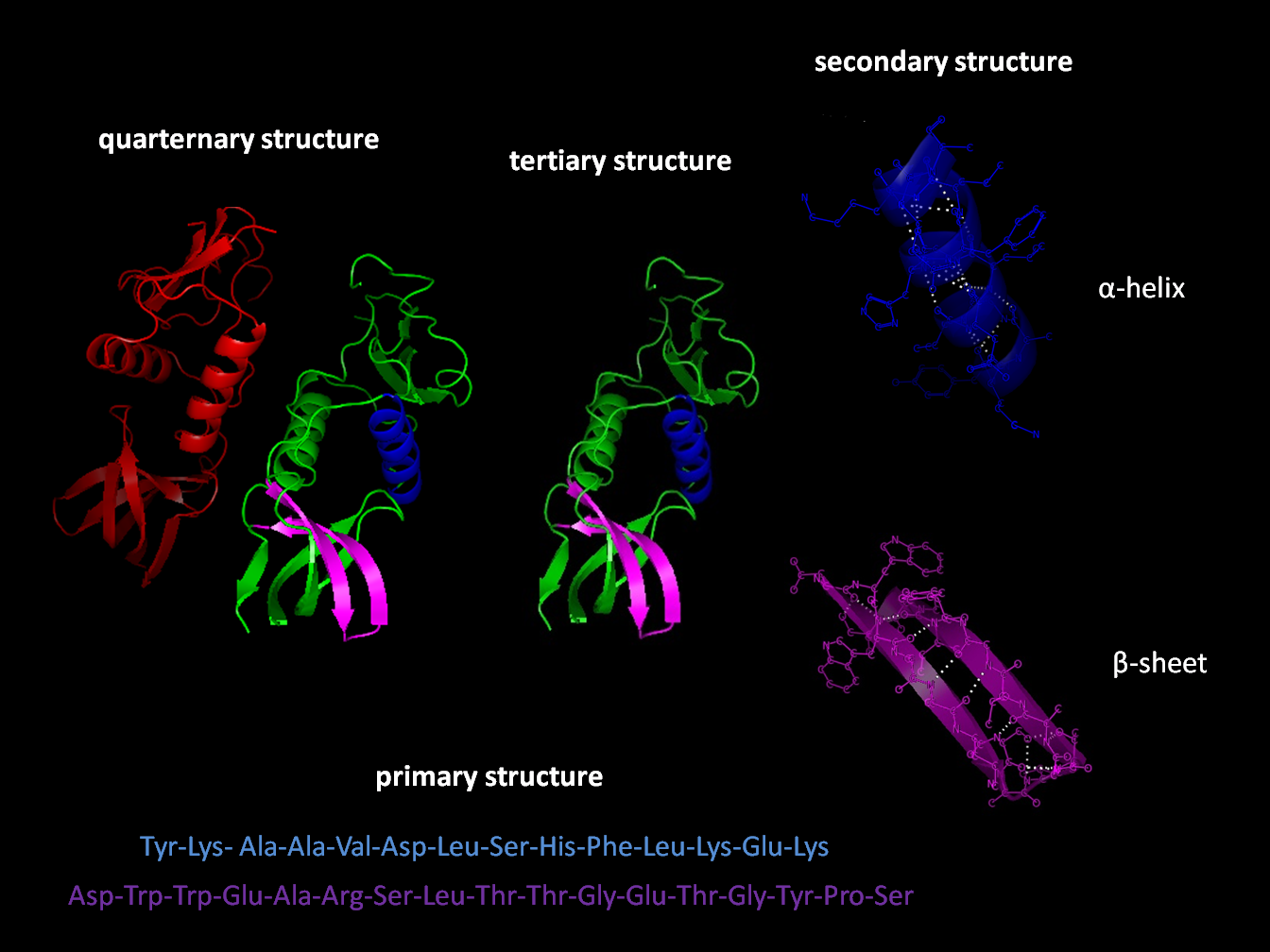
The primary structure of a protein, its linear amino-acid sequence, determines its native conformation. The specific amino acid residues and their position in the polypeptide chain are the determining factors for which portions of the protein fold closely together and form its three-dimensional conformation. The amino acid composition is not as important as the sequence. The essential fact of folding, however, remains that the amino acid sequence of each protein contains the information that specifies both the native structure and the pathway to attain that state. This is not to say that nearly identical amino acid sequences always fold similarly. Conformations differ based on environmental factors as well; similar proteins fold differently based on where they are found.
Formation of a secondary structure is the first step in the folding process that a protein takes to assume its native structure. Characteristic of secondary structure are the structures known as alpha helices and beta sheets that fold rapidly because they are stabilized by intramolecular hydrogen bonds, as was first characterized by Linus Pauling. Formation of intramolecular hydrogen bonds provides another important contribution to protein stability. α-helices are formed by hydrogen bonding of the backbone to form a spiral shape (refer to figure on the right). The β pleated sheet is a structure that forms with the backbone bending over itself to form the hydrogen bonds (as displayed in the figure to the left). The hydrogen bonds are between the amide hydrogen and carbonyl oxygen of the peptide bond. There exists anti-parallel β pleated sheets and parallel β pleated sheets where the stability of the hydrogen bonds is stronger in the anti-parallel β sheet as it hydrogen bonds with the ideal 180 degree angle compared to the slanted hydrogen bonds formed by parallel sheets.
Protein folding
Protein folding is the physical process by which a protein, after synthesis by a ribosome as a linear chain of amino acids, changes from an unstable random coil into a more ordered three-dimensional structure. This structure permits the protein to become biologically functional or active.
The folding of many proteins begins even during the translation of the polypeptide chain. The amino acids interact with each other to produce a well-defined three-dimensional structure, known as the protein's native state. This structure is determined by the amino-acid sequence or primary structure.
The correct three-dimensional structure is essential to function, although some parts of functional proteins may remain unfolded, indicating that protein dynamics are important. Failure to fold into a native structure generally produces inactive proteins, but in some instances, misfolded proteins have modified or toxic functionality. Several neurodegenerative and other diseases are believed to result from the accumulation of amyloid fibrils formed by misfolded proteins, the infectious varieties of which are known as prions. Many allergies are caused by the incorrect folding of some proteins because the immune system does not produce the antibodies for certain protein structures.
Denaturation of proteins is a process of transition from a folded to an unfolded state. It happens in cooking, burns, proteinopathies, and other contexts. Residual structure present, if any, in the supposedly unfolded state may form a folding initiation site and guide the subsequent folding reactions.
The duration of the folding process varies dramatically depending on the protein of interest. When studied outside the cell, the slowest folding proteins require many minutes or hours to fold, primarily due to proline isomerization, and must pass through a number of intermediate states, like checkpoints, before the process is complete. On the other hand, very small single-domain proteins with lengths of up to a hundred amino acids typically fold in a single step. Time scales of milliseconds are the norm, and the fastest known protein folding reactions are complete within a few microseconds. The folding time scale of a protein depends on its size, contact order, and circuit topology.
Understanding and simulating the protein folding process has been an important challenge for computational biology since the late 1960s.
The primary structure of a protein, its linear amino-acid sequence, determines its native conformation. The specific amino acid residues and their position in the polypeptide chain are the determining factors for which portions of the protein fold closely together and form its three-dimensional conformation. The amino acid composition is not as important as the sequence. The essential fact of folding, however, remains that the amino acid sequence of each protein contains the information that specifies both the native structure and the pathway to attain that state. This is not to say that nearly identical amino acid sequences always fold similarly. Conformations differ based on environmental factors as well; similar proteins fold differently based on where they are found.
Formation of a secondary structure is the first step in the folding process that a protein takes to assume its native structure. Characteristic of secondary structure are the structures known as alpha helices and beta sheets that fold rapidly because they are stabilized by intramolecular hydrogen bonds, as was first characterized by Linus Pauling. Formation of intramolecular hydrogen bonds provides another important contribution to protein stability. α-helices are formed by hydrogen bonding of the backbone to form a spiral shape (refer to figure on the right). The β pleated sheet is a structure that forms with the backbone bending over itself to form the hydrogen bonds (as displayed in the figure to the left). The hydrogen bonds are between the amide hydrogen and carbonyl oxygen of the peptide bond. There exists anti-parallel β pleated sheets and parallel β pleated sheets where the stability of the hydrogen bonds is stronger in the anti-parallel β sheet as it hydrogen bonds with the ideal 180 degree angle compared to the slanted hydrogen bonds formed by parallel sheets.
Recent media
Recent media