
Community hub
Recent from talks
Contribute something to knowledge base
Content stats: 0 posts, 0 articles, 1 media, 0 notes
Members stats: 0 subscribers, 0 contributors, 0 moderators, 0 supporters
Subscribers
Supporters
Contributors
Moderators
Hub AI
Eicosanoid AI simulator
(@Eicosanoid_simulator)
Hub AI
Eicosanoid AI simulator
(@Eicosanoid_simulator)
Eicosanoid
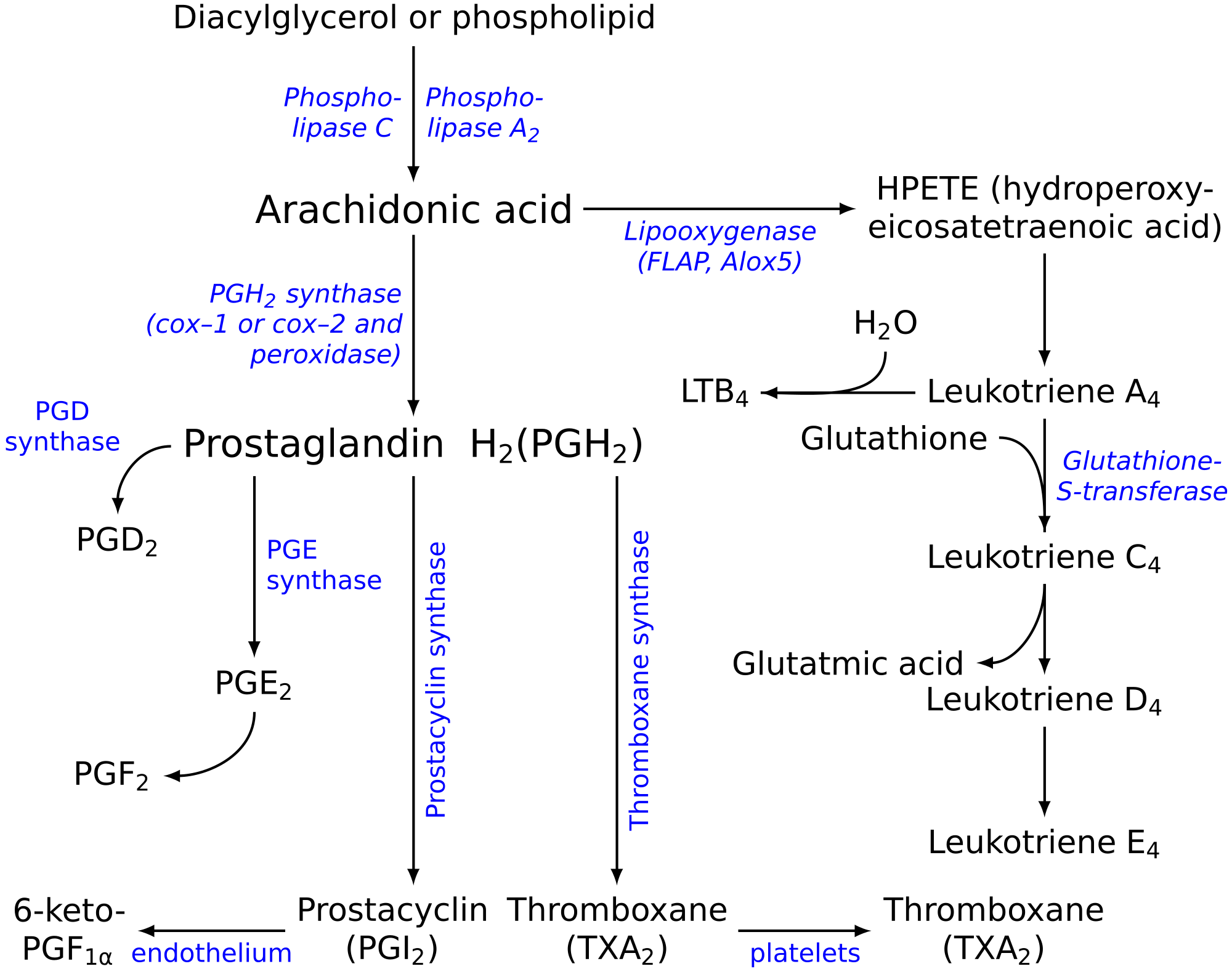
Eicosanoids are signaling molecules made by the enzymatic or non-enzymatic oxidation of arachidonic acid or other polyunsaturated fatty acids (PUFAs) that are, similar to arachidonic acid, around 20 carbon units in length. Eicosanoids are a sub-category of oxylipins, i.e. oxidized fatty acids of diverse carbon units in length, and are distinguished from other oxylipins by their overwhelming importance as cell signaling molecules. Eicosanoids function in diverse physiological systems and pathological processes such as: mounting or inhibiting inflammation, allergy, fever and other immune responses; regulating the abortion of pregnancy and normal childbirth; contributing to the perception of pain; regulating cell growth; controlling blood pressure; and modulating the regional flow of blood to tissues. In performing these roles, eicosanoids most often act as autocrine signaling agents to impact their cells of origin or as paracrine signaling agents to impact cells in the proximity of their cells of origin. Some eicosanoids, such as prostaglandins, may also have endocrine roles as hormones to influence the function of distant cells.
There are multiple subfamilies of eicosanoids, including most prominently the prostaglandins, thromboxanes, leukotrienes, lipoxins, resolvins, and eoxins. For each subfamily, there is the potential to have at least 4 separate series of metabolites, two series derived from the ω−6 PUFAs arachidonic and dihomo-gamma-linolenic acids, one series derived from the ω−3 PUFA eicosapentaenoic acid, and one series derived from the ω−9 PUFA mead acid. This subfamily distinction is important. Mammals, including humans, are unable to convert ω−6 into ω−3 PUFA. In consequence, tissue levels of the ω−6 and ω−3 PUFAs and their corresponding eicosanoid metabolites link directly to the amount of dietary ω−6 versus ω−3 PUFAs consumed. Since certain of the ω−6 and ω−3 PUFA series of metabolites have almost diametrically opposing physiological and pathological activities, it has often been suggested that the deleterious consequences associated with the consumption of ω−6 PUFA-rich diets reflects excessive production and activities of ω−6 PUFA-derived eicosanoids, while the beneficial effects associated with the consumption of ω−3 PUFA-rich diets reflect the excessive production and activities of ω−3 PUFA-derived eicosanoids. In this view, the opposing effects of ω−6 PUFA-derived and ω−3 PUFA-derived eicosanoids on key target cells underlie the detrimental and beneficial effects of ω−6 and ω−3 PUFA-rich diets on inflammation and allergy reactions, atherosclerosis, hypertension, cancer growth, and a host of other processes.
"Eicosanoid" (from Greek eicosa- 'twenty') is the collective term for straight-chain PUFAs (polyunsaturated fatty acids) of 20 carbon units in length that have been metabolized or otherwise converted to oxygen-containing products. The PUFA precursors to the eicosanoids include:
A particular eicosanoid is denoted by a four-character abbreviation, composed of:
The stereochemistry of the eicosanoid products formed may differ among the pathways. For prostaglandins, this is often indicated by Greek letters (e.g. PGF2α versus PGF2β). For hydroperoxy and hydroxy eicosanoids an S or R designates the chirality of their substituents (e.g. 5S-hydroxy-eicosateteraenoic acid [also termed 5(S)-, 5S-hydroxy-, and 5(S)-hydroxy-eicosatetraenoic acid] is given the trivial names of 5S-HETE, 5(S)-HETE, 5S-HETE, or 5(S)-HETE). Since eicosanoid-forming enzymes commonly make S isomer products either with marked preference or essentially exclusively, the use of S/R designations has often been dropped (e.g. 5S-HETE is 5-HETE). Nonetheless, certain eicosanoid-forming pathways do form R isomers and their S versus R isomeric products can exhibit dramatically different biological activities. Failing to specify S/R isomers can be misleading. Here, all hydroperoxy and hydroxy substituents have the S configuration unless noted otherwise.
Current usage limits the term eicosanoid to:
Hydroxyeicosatetraenoic acids, leukotrienes, eoxins and prostanoids are sometimes termed "classic eicosanoids".
In contrast to the classic eicosanoids, several other classes of PUFA metabolites have been termed 'novel', 'eicosanoid-like' or 'nonclassic eicosanoids'. These included the following classes:
Eicosanoid
Eicosanoids are signaling molecules made by the enzymatic or non-enzymatic oxidation of arachidonic acid or other polyunsaturated fatty acids (PUFAs) that are, similar to arachidonic acid, around 20 carbon units in length. Eicosanoids are a sub-category of oxylipins, i.e. oxidized fatty acids of diverse carbon units in length, and are distinguished from other oxylipins by their overwhelming importance as cell signaling molecules. Eicosanoids function in diverse physiological systems and pathological processes such as: mounting or inhibiting inflammation, allergy, fever and other immune responses; regulating the abortion of pregnancy and normal childbirth; contributing to the perception of pain; regulating cell growth; controlling blood pressure; and modulating the regional flow of blood to tissues. In performing these roles, eicosanoids most often act as autocrine signaling agents to impact their cells of origin or as paracrine signaling agents to impact cells in the proximity of their cells of origin. Some eicosanoids, such as prostaglandins, may also have endocrine roles as hormones to influence the function of distant cells.
There are multiple subfamilies of eicosanoids, including most prominently the prostaglandins, thromboxanes, leukotrienes, lipoxins, resolvins, and eoxins. For each subfamily, there is the potential to have at least 4 separate series of metabolites, two series derived from the ω−6 PUFAs arachidonic and dihomo-gamma-linolenic acids, one series derived from the ω−3 PUFA eicosapentaenoic acid, and one series derived from the ω−9 PUFA mead acid. This subfamily distinction is important. Mammals, including humans, are unable to convert ω−6 into ω−3 PUFA. In consequence, tissue levels of the ω−6 and ω−3 PUFAs and their corresponding eicosanoid metabolites link directly to the amount of dietary ω−6 versus ω−3 PUFAs consumed. Since certain of the ω−6 and ω−3 PUFA series of metabolites have almost diametrically opposing physiological and pathological activities, it has often been suggested that the deleterious consequences associated with the consumption of ω−6 PUFA-rich diets reflects excessive production and activities of ω−6 PUFA-derived eicosanoids, while the beneficial effects associated with the consumption of ω−3 PUFA-rich diets reflect the excessive production and activities of ω−3 PUFA-derived eicosanoids. In this view, the opposing effects of ω−6 PUFA-derived and ω−3 PUFA-derived eicosanoids on key target cells underlie the detrimental and beneficial effects of ω−6 and ω−3 PUFA-rich diets on inflammation and allergy reactions, atherosclerosis, hypertension, cancer growth, and a host of other processes.
"Eicosanoid" (from Greek eicosa- 'twenty') is the collective term for straight-chain PUFAs (polyunsaturated fatty acids) of 20 carbon units in length that have been metabolized or otherwise converted to oxygen-containing products. The PUFA precursors to the eicosanoids include:
A particular eicosanoid is denoted by a four-character abbreviation, composed of:
The stereochemistry of the eicosanoid products formed may differ among the pathways. For prostaglandins, this is often indicated by Greek letters (e.g. PGF2α versus PGF2β). For hydroperoxy and hydroxy eicosanoids an S or R designates the chirality of their substituents (e.g. 5S-hydroxy-eicosateteraenoic acid [also termed 5(S)-, 5S-hydroxy-, and 5(S)-hydroxy-eicosatetraenoic acid] is given the trivial names of 5S-HETE, 5(S)-HETE, 5S-HETE, or 5(S)-HETE). Since eicosanoid-forming enzymes commonly make S isomer products either with marked preference or essentially exclusively, the use of S/R designations has often been dropped (e.g. 5S-HETE is 5-HETE). Nonetheless, certain eicosanoid-forming pathways do form R isomers and their S versus R isomeric products can exhibit dramatically different biological activities. Failing to specify S/R isomers can be misleading. Here, all hydroperoxy and hydroxy substituents have the S configuration unless noted otherwise.
Current usage limits the term eicosanoid to:
Hydroxyeicosatetraenoic acids, leukotrienes, eoxins and prostanoids are sometimes termed "classic eicosanoids".
In contrast to the classic eicosanoids, several other classes of PUFA metabolites have been termed 'novel', 'eicosanoid-like' or 'nonclassic eicosanoids'. These included the following classes:
Recent media
Recent media