Recent from talks
Familial hypercholesterolemia
Knowledge base stats:
Talk channels stats:
Members stats:
Familial hypercholesterolemia
Familial hypercholesterolemia (FH) is a genetic disorder characterized by high cholesterol levels, specifically very high levels of low-density lipoprotein cholesterol (LDL cholesterol), in the blood and early cardiovascular diseases. The most common mutations diminish the number of functional LDL receptors in the liver or produce abnormal LDL receptors that never go to the cell surface to function properly (abnormal trafficking). Since the underlying body biochemistry is slightly different in individuals with FH, their high cholesterol levels are less responsive to the kinds of cholesterol control methods which are usually more effective in people without FH (such as dietary modification and statin tablets). Nevertheless, treatment (including higher statin doses and PCSK9 inhibitors) is usually effective.
FH is classified as a type 2 familial dyslipidemia. There are five types of familial dyslipidemia (not including subtypes), and each are classified from both the altered lipid profile and by the genetic abnormality. For example, high LDL (often due to LDL receptor defect) is type 2. Others include defects in chylomicron metabolism, triglyceride metabolism, and the metabolism of other cholesterol-containing particles, such as VLDL and IDL.
About 1 in 100 to 200 people have mutations in the LDLR gene that encodes the LDL receptor protein, which normally removes LDL from circulation, or the APOB gene that encodes apolipoprotein B (ApoB), the part of LDL particles that binds with LDL receptors. Mutations in other genes are rare but important to know, including gain-of-function mutations in the PCSK9 gene coding for the PCSK9 enzyme (which degrades LDL receptors), resulting in less LDL receptors available. PCSK9 mutations cause less than 5% of cases of FH according to most epidemiologic studies. People who have one abnormal copy (are heterozygous) of the LDLR gene may develop cardiovascular disease prematurely at the age of 30 to 40. Having two abnormal copies (being homozygous) may cause severe cardiovascular disease in childhood. Heterozygous FH is a common genetic disorder, inherited in an autosomal dominant pattern, occurring in 1:250 people in most countries; homozygous FH is much rarer, occurring in 1 in 300,000 people.[citation needed]
Heterozygous FH is normally treated with statins, bile acid sequestrants, or other lipid-lowering agents that lower cholesterol levels. New cases are generally offered genetic counseling. Homozygous FH often does not respond to medical therapy and may require other treatments, including LDL apheresis (removal of LDL in a method similar to dialysis) and occasionally liver transplantation.
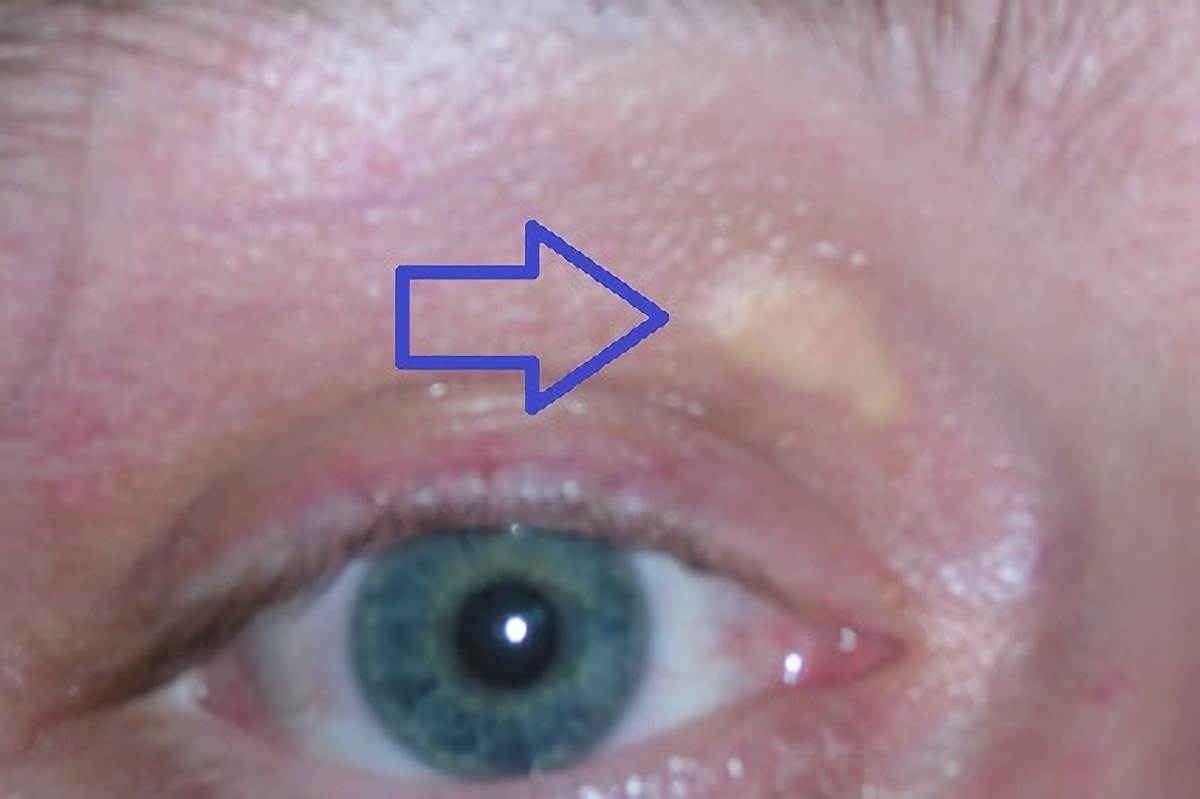
High cholesterol levels normally do not cause any symptoms. Yellow deposits of cholesterol-rich fat may be seen in various places on the body such as around the eyelids (known as xanthelasma palpebrarum), the outer margin of the iris (known as arcus senilis corneae), and in the tendons of the hands, elbows, knees, and feet, particularly the Achilles tendon (known as a tendon xanthoma).
Accelerated deposition of cholesterol in the walls of arteries leads to atherosclerosis, the underlying cause of cardiovascular disease. The most common problem in FH is the development of coronary artery disease (atherosclerosis of the coronary arteries that supply the heart) at a much younger age than would be expected in the general population. This may lead to angina pectoris (chest pain or tightness on exertion) or heart attacks. Less commonly, arteries of the brain are affected; this may lead to transient ischemic attacks (brief episodes of weakness on one side of the body or inability to talk) or occasionally stroke. Peripheral artery occlusive disease (obstruction of the arteries of the legs) occurs mainly in people with FH who smoke; this can cause pain in the calf muscles during walking that resolves with rest (intermittent claudication) and problems due to a decreased blood supply to the feet (such as gangrene). Atherosclerosis risk is increased further with age and in those who smoke, have diabetes, high blood pressure and a family history of cardiovascular disease.
Approximately 85% of individuals with this disorder have not been diagnosed and consequently are not receiving lipid-lowering treatments. Physical examination findings can help a physician make the diagnosis of FH. Tendon xanthomas are seen in 20-40% of individuals with FH and are pathognomonic for the condition. A xanthelasma or corneal arcus may also be seen. These common signs are supportive of the diagnosis but are non-specific findings.
Cholesterol levels may be determined as part of health screening for health insurance or occupational health, when the external physical signs such as xanthelasma, xanthoma, arcus are noticed, symptoms of cardiovascular disease develop, or a family member has been found to have FH. A pattern compatible with hyperlipoproteinemia type IIa on the Fredrickson classification is typically found: raised level of total cholesterol, markedly raised level of low-density lipoprotein (LDL), normal level of high-density lipoprotein (HDL), and normal level of triglycerides. Total cholesterol levels of 350–550 mg/dL are typical of heterozygous FH while total cholesterol levels of 650–1000 mg/dL are typical of homozygous FH. The LDL is typically above the 75th percentile, that is, 75% of the healthy population would have a lower LDL level. Cholesterol levels can be drastically higher in people with FH who are also obese.
Hub AI
Familial hypercholesterolemia AI simulator
(@Familial hypercholesterolemia_simulator)
Familial hypercholesterolemia
Familial hypercholesterolemia (FH) is a genetic disorder characterized by high cholesterol levels, specifically very high levels of low-density lipoprotein cholesterol (LDL cholesterol), in the blood and early cardiovascular diseases. The most common mutations diminish the number of functional LDL receptors in the liver or produce abnormal LDL receptors that never go to the cell surface to function properly (abnormal trafficking). Since the underlying body biochemistry is slightly different in individuals with FH, their high cholesterol levels are less responsive to the kinds of cholesterol control methods which are usually more effective in people without FH (such as dietary modification and statin tablets). Nevertheless, treatment (including higher statin doses and PCSK9 inhibitors) is usually effective.
FH is classified as a type 2 familial dyslipidemia. There are five types of familial dyslipidemia (not including subtypes), and each are classified from both the altered lipid profile and by the genetic abnormality. For example, high LDL (often due to LDL receptor defect) is type 2. Others include defects in chylomicron metabolism, triglyceride metabolism, and the metabolism of other cholesterol-containing particles, such as VLDL and IDL.
About 1 in 100 to 200 people have mutations in the LDLR gene that encodes the LDL receptor protein, which normally removes LDL from circulation, or the APOB gene that encodes apolipoprotein B (ApoB), the part of LDL particles that binds with LDL receptors. Mutations in other genes are rare but important to know, including gain-of-function mutations in the PCSK9 gene coding for the PCSK9 enzyme (which degrades LDL receptors), resulting in less LDL receptors available. PCSK9 mutations cause less than 5% of cases of FH according to most epidemiologic studies. People who have one abnormal copy (are heterozygous) of the LDLR gene may develop cardiovascular disease prematurely at the age of 30 to 40. Having two abnormal copies (being homozygous) may cause severe cardiovascular disease in childhood. Heterozygous FH is a common genetic disorder, inherited in an autosomal dominant pattern, occurring in 1:250 people in most countries; homozygous FH is much rarer, occurring in 1 in 300,000 people.[citation needed]
Heterozygous FH is normally treated with statins, bile acid sequestrants, or other lipid-lowering agents that lower cholesterol levels. New cases are generally offered genetic counseling. Homozygous FH often does not respond to medical therapy and may require other treatments, including LDL apheresis (removal of LDL in a method similar to dialysis) and occasionally liver transplantation.
High cholesterol levels normally do not cause any symptoms. Yellow deposits of cholesterol-rich fat may be seen in various places on the body such as around the eyelids (known as xanthelasma palpebrarum), the outer margin of the iris (known as arcus senilis corneae), and in the tendons of the hands, elbows, knees, and feet, particularly the Achilles tendon (known as a tendon xanthoma).
Accelerated deposition of cholesterol in the walls of arteries leads to atherosclerosis, the underlying cause of cardiovascular disease. The most common problem in FH is the development of coronary artery disease (atherosclerosis of the coronary arteries that supply the heart) at a much younger age than would be expected in the general population. This may lead to angina pectoris (chest pain or tightness on exertion) or heart attacks. Less commonly, arteries of the brain are affected; this may lead to transient ischemic attacks (brief episodes of weakness on one side of the body or inability to talk) or occasionally stroke. Peripheral artery occlusive disease (obstruction of the arteries of the legs) occurs mainly in people with FH who smoke; this can cause pain in the calf muscles during walking that resolves with rest (intermittent claudication) and problems due to a decreased blood supply to the feet (such as gangrene). Atherosclerosis risk is increased further with age and in those who smoke, have diabetes, high blood pressure and a family history of cardiovascular disease.
Approximately 85% of individuals with this disorder have not been diagnosed and consequently are not receiving lipid-lowering treatments. Physical examination findings can help a physician make the diagnosis of FH. Tendon xanthomas are seen in 20-40% of individuals with FH and are pathognomonic for the condition. A xanthelasma or corneal arcus may also be seen. These common signs are supportive of the diagnosis but are non-specific findings.
Cholesterol levels may be determined as part of health screening for health insurance or occupational health, when the external physical signs such as xanthelasma, xanthoma, arcus are noticed, symptoms of cardiovascular disease develop, or a family member has been found to have FH. A pattern compatible with hyperlipoproteinemia type IIa on the Fredrickson classification is typically found: raised level of total cholesterol, markedly raised level of low-density lipoprotein (LDL), normal level of high-density lipoprotein (HDL), and normal level of triglycerides. Total cholesterol levels of 350–550 mg/dL are typical of heterozygous FH while total cholesterol levels of 650–1000 mg/dL are typical of homozygous FH. The LDL is typically above the 75th percentile, that is, 75% of the healthy population would have a lower LDL level. Cholesterol levels can be drastically higher in people with FH who are also obese.
Recent media