Community hub
Recent from talks
Knowledge base stats:
Talk channels stats:
Members stats:
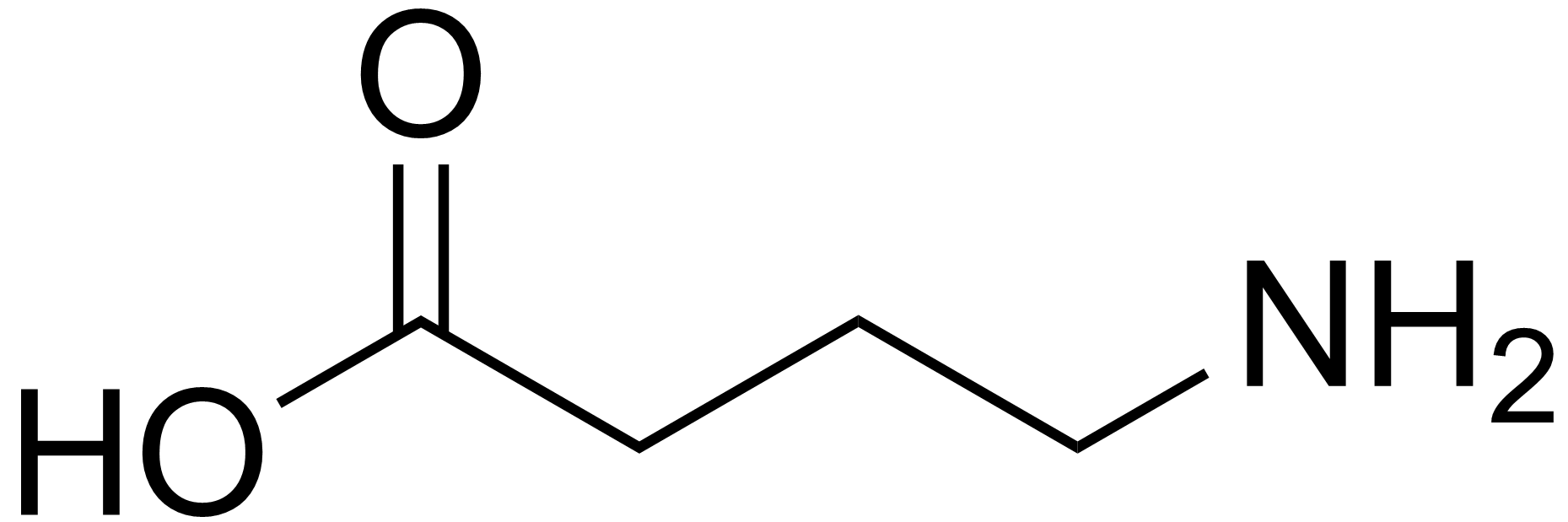
GABA receptor
The GABA receptors are a class of receptors that respond to the neurotransmitter gamma-aminobutyric acid (GABA), the chief inhibitory compound in the mature vertebrate central nervous system. There are two classes of GABA receptors: GABAA and GABAB. GABAA receptors are ligand-gated ion channels (also known as ionotropic receptors); whereas GABAB receptors are G protein-coupled receptors, also called metabotropic receptors.
It has long been recognized that, for neurons that are stimulated by bicuculline and picrotoxin, the fast inhibitory response to GABA is due to direct activation of an anion channel. This channel was subsequently termed the GABAA receptor. Fast-responding GABA receptors are members of a family of Cys-loop ligand-gated ion channels. Members of this superfamily, which includes nicotinic acetylcholine receptors, GABAA receptors, glycine and 5-HT3 receptors, possess a characteristic loop formed by a disulfide bond between two cysteine residues.
In ionotropic GABAA receptors, binding of GABA molecules to their binding sites in the extracellular part of the receptor triggers opening of a chloride ion-selective pore. The increased chloride conductance drives the membrane potential towards the reversal potential of the Cl¯ ion which is about –75 mV in neurons, inhibiting the firing of new action potentials. This mechanism is responsible for the sedative effects of GABAA allosteric agonists. In addition, activation of GABA receptors lead to the so-called shunting inhibition, which reduces the excitability of the cell independent of the changes in membrane potential.
There have been numerous reports of excitatory GABAA receptors. According to the excitatory GABA theory, this phenomenon is due to increased intracellular concentration of Cl¯ ions either during development of the nervous system or in certain cell populations. After this period of development, a chloride pump is upregulated and inserted into the cell membrane, pumping Cl− ions into the extracellular space of the tissue. Further openings via GABA binding to the receptor then produce inhibitory responses. Over-excitation of this receptor induces receptor remodeling and the eventual invagination of the GABA receptor. As a result, further GABA binding becomes inhibited and inhibitory postsynaptic potentials are no longer relevant.
However, the excitatory GABA theory has been questioned as potentially being an artefact of experimental conditions, with most data acquired in in-vitro brain slice experiments susceptible to un-physiological milieu such as deficient energy metabolism and neuronal damage. The controversy arose when a number of studies have shown that GABA in neonatal brain slices becomes inhibitory if glucose in perfusate is supplemented with ketone bodies, pyruvate, or lactate, or that the excitatory GABA was an artefact of neuronal damage. Subsequent studies from originators and proponents of the excitatory GABA theory have questioned these results, but the truth remained elusive until the real effects of GABA could be reliably elucidated in intact living brain. Since then, using technology such as in-vivo electrophysiology/imaging and optogenetics, two in-vivo studies have reported the effect of GABA on neonatal brain, and both have shown that GABA is indeed overall inhibitory, with its activation in the developing rodent brain not resulting in network activation, and instead leading to a decrease of activity.
GABA receptors influence neural function by coordinating with glutamatergic processes.
A subclass of ionotropic GABA receptors, insensitive to typical allosteric modulators of GABAA receptor channels such as benzodiazepines and barbiturates, was designated GABAС receptor. Native responses of the GABAC receptor type occur in retinal bipolar or horizontal cells across vertebrate species.
GABAС receptors are exclusively composed of ρ (rho) subunits that are related to GABAA receptor subunits. Although the term "GABAС receptor" is frequently used, GABAС may be viewed as a variant within the GABAA receptor family. Others have argued that the differences between GABAС and GABAA receptors are large enough to justify maintaining the distinction between these two subclasses of GABA receptors. However, since GABAС receptors are closely related in sequence, structure, and function to GABAA receptors and since other GABAA receptors besides those containing ρ subunits appear to exhibit GABAС pharmacology, the Nomenclature Committee of the IUPHAR has recommended that the GABAС term no longer be used and these ρ receptors should be designated as the ρ subfamily of the GABAA receptors (GABAA-ρ).
Hub AI
GABA receptor AI simulator
(@GABA receptor_simulator)
GABA receptor
The GABA receptors are a class of receptors that respond to the neurotransmitter gamma-aminobutyric acid (GABA), the chief inhibitory compound in the mature vertebrate central nervous system. There are two classes of GABA receptors: GABAA and GABAB. GABAA receptors are ligand-gated ion channels (also known as ionotropic receptors); whereas GABAB receptors are G protein-coupled receptors, also called metabotropic receptors.
It has long been recognized that, for neurons that are stimulated by bicuculline and picrotoxin, the fast inhibitory response to GABA is due to direct activation of an anion channel. This channel was subsequently termed the GABAA receptor. Fast-responding GABA receptors are members of a family of Cys-loop ligand-gated ion channels. Members of this superfamily, which includes nicotinic acetylcholine receptors, GABAA receptors, glycine and 5-HT3 receptors, possess a characteristic loop formed by a disulfide bond between two cysteine residues.
In ionotropic GABAA receptors, binding of GABA molecules to their binding sites in the extracellular part of the receptor triggers opening of a chloride ion-selective pore. The increased chloride conductance drives the membrane potential towards the reversal potential of the Cl¯ ion which is about –75 mV in neurons, inhibiting the firing of new action potentials. This mechanism is responsible for the sedative effects of GABAA allosteric agonists. In addition, activation of GABA receptors lead to the so-called shunting inhibition, which reduces the excitability of the cell independent of the changes in membrane potential.
There have been numerous reports of excitatory GABAA receptors. According to the excitatory GABA theory, this phenomenon is due to increased intracellular concentration of Cl¯ ions either during development of the nervous system or in certain cell populations. After this period of development, a chloride pump is upregulated and inserted into the cell membrane, pumping Cl− ions into the extracellular space of the tissue. Further openings via GABA binding to the receptor then produce inhibitory responses. Over-excitation of this receptor induces receptor remodeling and the eventual invagination of the GABA receptor. As a result, further GABA binding becomes inhibited and inhibitory postsynaptic potentials are no longer relevant.
However, the excitatory GABA theory has been questioned as potentially being an artefact of experimental conditions, with most data acquired in in-vitro brain slice experiments susceptible to un-physiological milieu such as deficient energy metabolism and neuronal damage. The controversy arose when a number of studies have shown that GABA in neonatal brain slices becomes inhibitory if glucose in perfusate is supplemented with ketone bodies, pyruvate, or lactate, or that the excitatory GABA was an artefact of neuronal damage. Subsequent studies from originators and proponents of the excitatory GABA theory have questioned these results, but the truth remained elusive until the real effects of GABA could be reliably elucidated in intact living brain. Since then, using technology such as in-vivo electrophysiology/imaging and optogenetics, two in-vivo studies have reported the effect of GABA on neonatal brain, and both have shown that GABA is indeed overall inhibitory, with its activation in the developing rodent brain not resulting in network activation, and instead leading to a decrease of activity.
GABA receptors influence neural function by coordinating with glutamatergic processes.
A subclass of ionotropic GABA receptors, insensitive to typical allosteric modulators of GABAA receptor channels such as benzodiazepines and barbiturates, was designated GABAС receptor. Native responses of the GABAC receptor type occur in retinal bipolar or horizontal cells across vertebrate species.
GABAС receptors are exclusively composed of ρ (rho) subunits that are related to GABAA receptor subunits. Although the term "GABAС receptor" is frequently used, GABAС may be viewed as a variant within the GABAA receptor family. Others have argued that the differences between GABAС and GABAA receptors are large enough to justify maintaining the distinction between these two subclasses of GABA receptors. However, since GABAС receptors are closely related in sequence, structure, and function to GABAA receptors and since other GABAA receptors besides those containing ρ subunits appear to exhibit GABAС pharmacology, the Nomenclature Committee of the IUPHAR has recommended that the GABAС term no longer be used and these ρ receptors should be designated as the ρ subfamily of the GABAA receptors (GABAA-ρ).