Recent from talks
Knowledge base stats:
Talk channels stats:
Members stats:
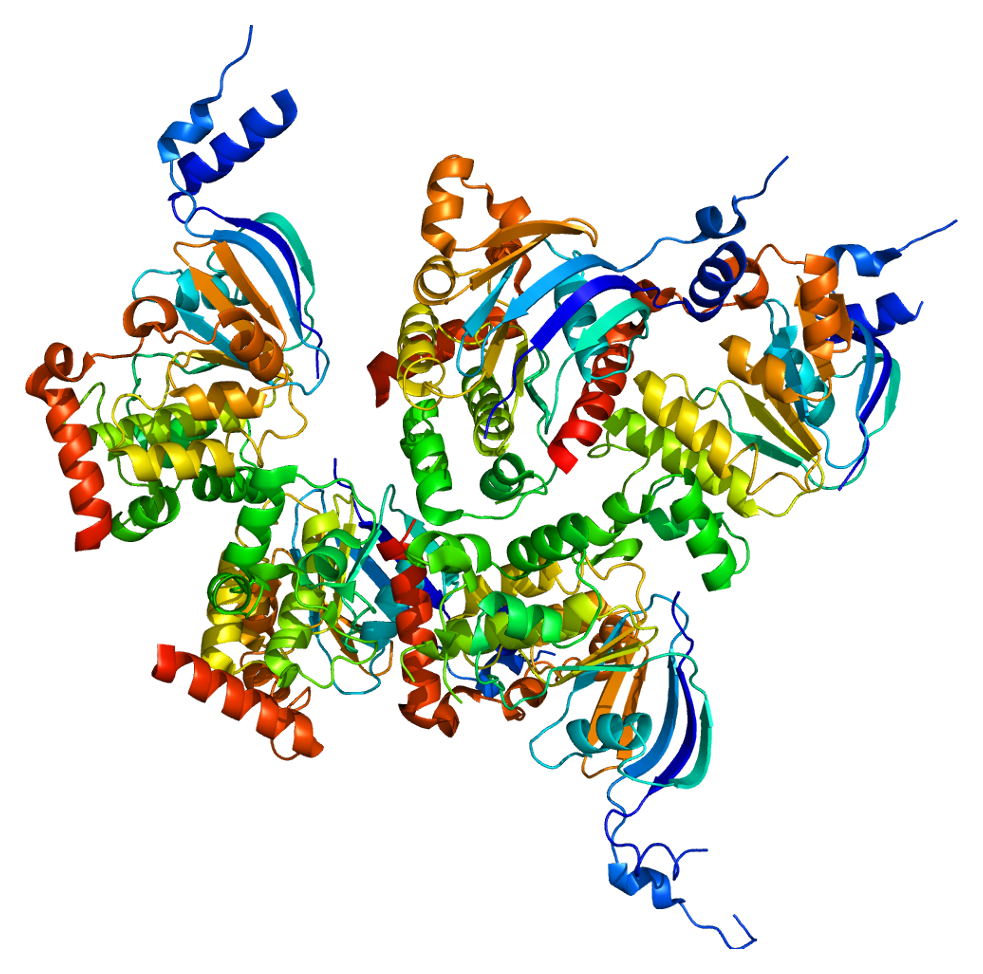
Cystic fibrosis transmembrane conductance regulator
Cystic fibrosis transmembrane conductance regulator (CFTR) is a membrane protein and anion channel in vertebrates that is encoded by the CFTR gene.
Geneticist Lap-Chee Tsui and his team identified the CFTR gene in 1989 as the gene linked with CF (cystic fibrosis).
The CFTR gene codes for an ABC transporter-class ion channel protein that conducts chloride and bicarbonate ions across epithelial cell membranes. Mutations of the CFTR gene affecting anion channel function lead to dysregulation of epithelial lining fluid (mucus) transport in the lung, pancreas and other organs, resulting in cystic fibrosis. Complications include thickened mucus in the lungs with frequent respiratory infections, and pancreatic insufficiency giving rise to malnutrition and diabetes. These conditions lead to chronic disability and reduced life expectancy. In male patients, the progressive obstruction and destruction of the developing vas deferens (spermatic cord) and epididymis appear to result from abnormal intraluminal secretions, causing congenital absence of the vas deferens and male infertility, and found associated with an imbalance of fatty acids.
The CFTR is found in the epithelial cells of many organs including the lung, liver, pancreas, digestive tract, and the female reproductive tract and male reproductive tract including the testis, Sertoli cells, spermatozoa. epididymis, and the vas deferens.
In the airways of the lung, CFTR is most highly expressed by rare specialized cells called pulmonary ionocytes. In the skin, CFTR is strongly expressed in the sebaceous and eccrine sweat glands. In the eccrine glands, CFTR is located on the apical membrane of the epithelial cells that make up the duct of these sweat glands.
Normally, the protein allows movement of chloride, bicarbonate and thiocyanate ions (with a negative charge) out of an epithelial cell into the airway surface liquid and mucus. Positively charged sodium ions follow passively, increasing the total electrolyte concentration in the mucus, resulting in the movement of water out of the cell via osmosis.
In epithelial cells with motile cilia lining the bronchus and the oviduct, CFTR is located on the apical cell membrane but not on cilia. In contrast, ENaC (Epithelial sodium channel) is located along the entire length of the cilia.
In sweat glands, defective CFTR results in reduced transport of sodium chloride and sodium thiocyanate in the resorptive duct and therefore saltier sweat. This is the basis of a clinically important sweat test for cystic fibrosis often used diagnostically with genetic screening.
Hub AI
Cystic fibrosis transmembrane conductance regulator AI simulator
(@Cystic fibrosis transmembrane conductance regulator_simulator)
Cystic fibrosis transmembrane conductance regulator
Cystic fibrosis transmembrane conductance regulator (CFTR) is a membrane protein and anion channel in vertebrates that is encoded by the CFTR gene.
Geneticist Lap-Chee Tsui and his team identified the CFTR gene in 1989 as the gene linked with CF (cystic fibrosis).
The CFTR gene codes for an ABC transporter-class ion channel protein that conducts chloride and bicarbonate ions across epithelial cell membranes. Mutations of the CFTR gene affecting anion channel function lead to dysregulation of epithelial lining fluid (mucus) transport in the lung, pancreas and other organs, resulting in cystic fibrosis. Complications include thickened mucus in the lungs with frequent respiratory infections, and pancreatic insufficiency giving rise to malnutrition and diabetes. These conditions lead to chronic disability and reduced life expectancy. In male patients, the progressive obstruction and destruction of the developing vas deferens (spermatic cord) and epididymis appear to result from abnormal intraluminal secretions, causing congenital absence of the vas deferens and male infertility, and found associated with an imbalance of fatty acids.
The CFTR is found in the epithelial cells of many organs including the lung, liver, pancreas, digestive tract, and the female reproductive tract and male reproductive tract including the testis, Sertoli cells, spermatozoa. epididymis, and the vas deferens.
In the airways of the lung, CFTR is most highly expressed by rare specialized cells called pulmonary ionocytes. In the skin, CFTR is strongly expressed in the sebaceous and eccrine sweat glands. In the eccrine glands, CFTR is located on the apical membrane of the epithelial cells that make up the duct of these sweat glands.
Normally, the protein allows movement of chloride, bicarbonate and thiocyanate ions (with a negative charge) out of an epithelial cell into the airway surface liquid and mucus. Positively charged sodium ions follow passively, increasing the total electrolyte concentration in the mucus, resulting in the movement of water out of the cell via osmosis.
In epithelial cells with motile cilia lining the bronchus and the oviduct, CFTR is located on the apical cell membrane but not on cilia. In contrast, ENaC (Epithelial sodium channel) is located along the entire length of the cilia.
In sweat glands, defective CFTR results in reduced transport of sodium chloride and sodium thiocyanate in the resorptive duct and therefore saltier sweat. This is the basis of a clinically important sweat test for cystic fibrosis often used diagnostically with genetic screening.