Recent from talks
Cyclohexane conformation
Knowledge base stats:
Talk channels stats:
Members stats:
Cyclohexane conformation
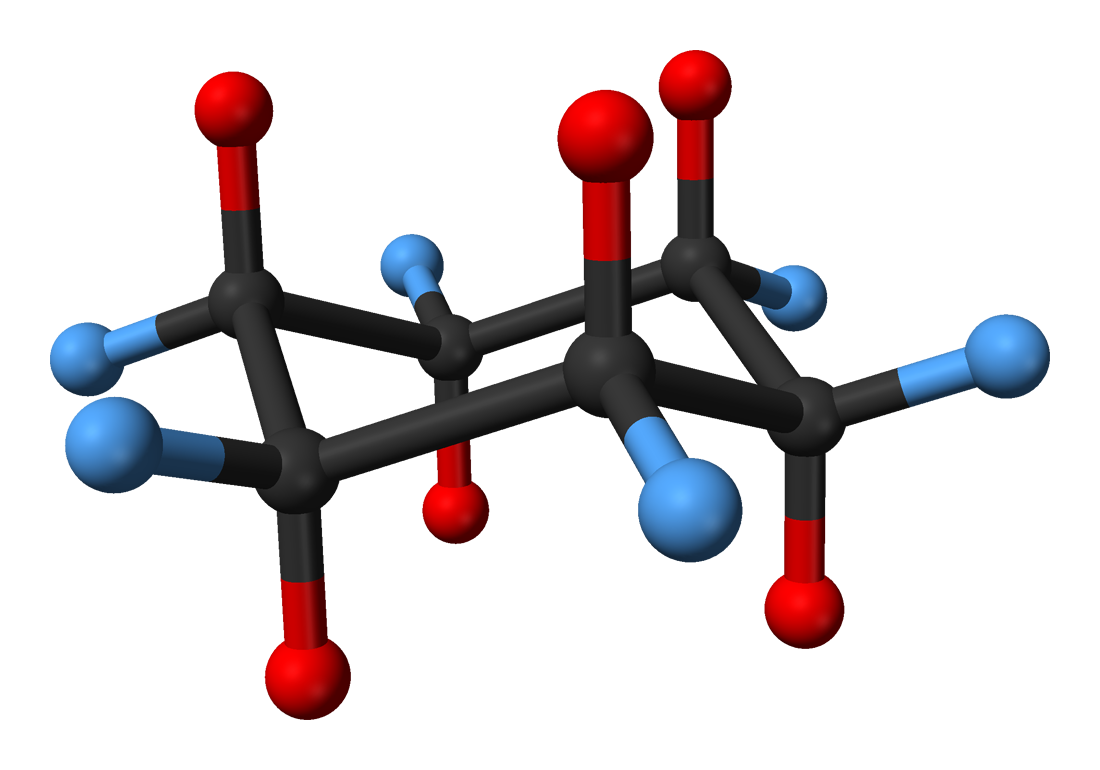
Cyclohexane conformations are any of several three-dimensional shapes adopted by cyclohexane. Because many compounds feature structurally similar six-membered rings, the structure and dynamics of cyclohexane are important prototypes of a wide range of compounds.
The internal angles of a regular, flat hexagon are 120°, while the preferred angle between successive bonds in a carbon chain is about 109.5°, the tetrahedral angle (the arc cosine of −1/3). Therefore, the cyclohexane ring tends to assume non-planar (warped) conformations, which have all angles closer to 109.5° and therefore a lower strain energy than the flat hexagonal shape.
Consider the carbon atoms numbered from 1 to 6 around the ring. If we hold carbon atoms 1, 2, and 3 stationary, with the correct bond lengths and the tetrahedral angle between the two bonds, and then continue by adding carbon atoms 4, 5, and 6 with the correct bond length and the tetrahedral angle, we can vary the three dihedral angles for the sequences (1,2,3,4), (2,3,4,5), and (3,4,5,6). The next bond, from atom 6, is also oriented by a dihedral angle, so we have four degrees of freedom. But that last bond has to end at the position of atom 1, which imposes three conditions in three-dimensional space. If the bond angle in the chain (6,1,2) should also be the tetrahedral angle then we have four conditions. Normally this would mean that there are no degrees of freedom of conformation, giving a finite number of solutions. With atoms 1, 2, and 3 fixed, there are two solutions, called chair (depending on whether the dihedral angle for (1,2,3,4) is positive or negative), but it turns out that there is also a continuum of solutions, a topological circle where angle strain is zero, including the twist boat and the boat conformations. All the conformations on this continuum have a twofold axis of symmetry running through the ring, whereas the chair conformations do not (they have D3d symmetry, with a threefold axis running through the ring). It is because of the symmetry of the conformations on this continuum that it is possible to satisfy all four constraints with a range of dihedral angles at (1,2,3,4). On this continuum the energy varies because of Pitzer strain related to the dihedral angles. The twist-boat has a lower energy than the boat. In order to go from the chair conformation to a twist-boat conformation or the other chair conformation, bond angles have to be changed, leading to a high-energy half-chair conformation. So the relative energies are: chair < twist-boat < boat < half-chair with chair being the most stable and half-chair the least. All relative conformational energies are shown below. At room temperature the molecule can easily move among these conformations, but only chair and twist-boat can be isolated in pure form, because the others are not at local energy minima.
The boat and twist-boat conformations, as said, lie along a continuum of zero angle strain. If there are substituents that allow the different carbon atoms to be distinguished, then this continuum is like a circle with six boat conformations and six twist-boat conformations between them, three "right-handed" and three "left-handed". (Which should be called right-handed is unimportant.) But if the carbon atoms are indistinguishable, as in cyclohexane itself, then moving along the continuum takes the molecule from the boat form to a "right-handed" twist-boat, and then back to the same boat form (with a permutation of the carbon atoms), then to a "left-handed" twist-boat, and then back again to the achiral boat. The passage from boat → right-twist-boat → boat → left-twist-boat → boat constitutes a full pseudorotation.
Another way to compare the stability within two molecules of cyclohexane in the same conformation is to evaluate the number of coplanar carbons in each molecule. Coplanar carbons are carbons that are all on the same plane. Increasing the number of coplanar carbons increases the number of eclipsing substituents. This increases the overall torsional strain and decreases the stability of the conformation. Cyclohexane diminishes the torsional strain from eclipsing substituents through adopting a conformation with fewer coplanar carbons. For example, if a half-chair conformation contains four coplanar carbons and another half-chair conformation contains five coplanar carbons, the conformation with four coplanar carbons will be more stable.
The different conformations are called "conformers", a blend of the words "conformation" and "isomer".
The chair conformation is the most stable conformer. At 298 K (25 °C), 99.99% of all molecules in a cyclohexane solution adopt this conformation.
The C–C ring of the chair conformation has the same shape as the 6-membered rings in the diamond cubic lattice. This can be modeled as follows. Consider a carbon atom to be a point with four half-bonds sticking out towards the vertices of a tetrahedron. Place it on a flat surface with one half-bond pointing straight up. Looking from directly above, the other three half-bonds will appear to point outwards towards the vertices of an equilateral triangle, so the bonds will appear to have an angle of 120° between them. Arrange six such atoms above the surface so that these 120° angles form a regular hexagon. Reflecting three of the atoms to be below the surface yields the desired geometry.
Hub AI
Cyclohexane conformation AI simulator
(@Cyclohexane conformation_simulator)
Cyclohexane conformation
Cyclohexane conformations are any of several three-dimensional shapes adopted by cyclohexane. Because many compounds feature structurally similar six-membered rings, the structure and dynamics of cyclohexane are important prototypes of a wide range of compounds.
The internal angles of a regular, flat hexagon are 120°, while the preferred angle between successive bonds in a carbon chain is about 109.5°, the tetrahedral angle (the arc cosine of −1/3). Therefore, the cyclohexane ring tends to assume non-planar (warped) conformations, which have all angles closer to 109.5° and therefore a lower strain energy than the flat hexagonal shape.
Consider the carbon atoms numbered from 1 to 6 around the ring. If we hold carbon atoms 1, 2, and 3 stationary, with the correct bond lengths and the tetrahedral angle between the two bonds, and then continue by adding carbon atoms 4, 5, and 6 with the correct bond length and the tetrahedral angle, we can vary the three dihedral angles for the sequences (1,2,3,4), (2,3,4,5), and (3,4,5,6). The next bond, from atom 6, is also oriented by a dihedral angle, so we have four degrees of freedom. But that last bond has to end at the position of atom 1, which imposes three conditions in three-dimensional space. If the bond angle in the chain (6,1,2) should also be the tetrahedral angle then we have four conditions. Normally this would mean that there are no degrees of freedom of conformation, giving a finite number of solutions. With atoms 1, 2, and 3 fixed, there are two solutions, called chair (depending on whether the dihedral angle for (1,2,3,4) is positive or negative), but it turns out that there is also a continuum of solutions, a topological circle where angle strain is zero, including the twist boat and the boat conformations. All the conformations on this continuum have a twofold axis of symmetry running through the ring, whereas the chair conformations do not (they have D3d symmetry, with a threefold axis running through the ring). It is because of the symmetry of the conformations on this continuum that it is possible to satisfy all four constraints with a range of dihedral angles at (1,2,3,4). On this continuum the energy varies because of Pitzer strain related to the dihedral angles. The twist-boat has a lower energy than the boat. In order to go from the chair conformation to a twist-boat conformation or the other chair conformation, bond angles have to be changed, leading to a high-energy half-chair conformation. So the relative energies are: chair < twist-boat < boat < half-chair with chair being the most stable and half-chair the least. All relative conformational energies are shown below. At room temperature the molecule can easily move among these conformations, but only chair and twist-boat can be isolated in pure form, because the others are not at local energy minima.
The boat and twist-boat conformations, as said, lie along a continuum of zero angle strain. If there are substituents that allow the different carbon atoms to be distinguished, then this continuum is like a circle with six boat conformations and six twist-boat conformations between them, three "right-handed" and three "left-handed". (Which should be called right-handed is unimportant.) But if the carbon atoms are indistinguishable, as in cyclohexane itself, then moving along the continuum takes the molecule from the boat form to a "right-handed" twist-boat, and then back to the same boat form (with a permutation of the carbon atoms), then to a "left-handed" twist-boat, and then back again to the achiral boat. The passage from boat → right-twist-boat → boat → left-twist-boat → boat constitutes a full pseudorotation.
Another way to compare the stability within two molecules of cyclohexane in the same conformation is to evaluate the number of coplanar carbons in each molecule. Coplanar carbons are carbons that are all on the same plane. Increasing the number of coplanar carbons increases the number of eclipsing substituents. This increases the overall torsional strain and decreases the stability of the conformation. Cyclohexane diminishes the torsional strain from eclipsing substituents through adopting a conformation with fewer coplanar carbons. For example, if a half-chair conformation contains four coplanar carbons and another half-chair conformation contains five coplanar carbons, the conformation with four coplanar carbons will be more stable.
The different conformations are called "conformers", a blend of the words "conformation" and "isomer".
The chair conformation is the most stable conformer. At 298 K (25 °C), 99.99% of all molecules in a cyclohexane solution adopt this conformation.
The C–C ring of the chair conformation has the same shape as the 6-membered rings in the diamond cubic lattice. This can be modeled as follows. Consider a carbon atom to be a point with four half-bonds sticking out towards the vertices of a tetrahedron. Place it on a flat surface with one half-bond pointing straight up. Looking from directly above, the other three half-bonds will appear to point outwards towards the vertices of an equilateral triangle, so the bonds will appear to have an angle of 120° between them. Arrange six such atoms above the surface so that these 120° angles form a regular hexagon. Reflecting three of the atoms to be below the surface yields the desired geometry.
Recent media