Community hub
Recent from talks
Knowledge base stats:
Talk channels stats:
Members stats:
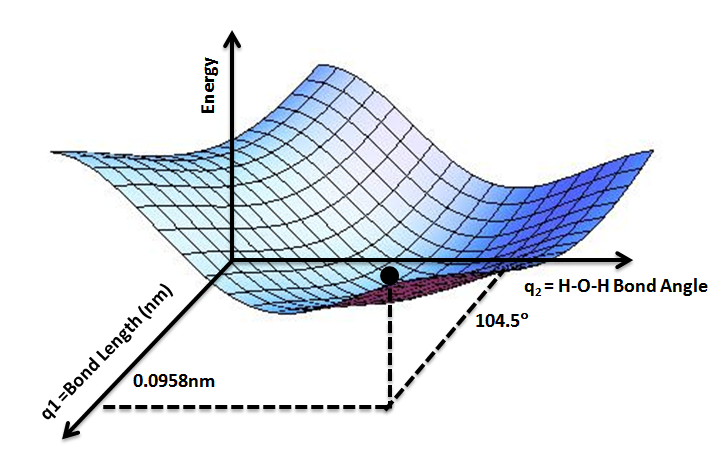
Potential energy surface
A potential energy surface (PES) or energy landscape describes the energy of a system, especially a collection of atoms, in terms of certain parameters, normally the positions of the atoms. The surface might define the energy as a function of one or more coordinates; if there is only one coordinate, the surface is called a potential energy curve or energy profile. An example is the Morse/Long-range potential.
It is helpful to use the analogy of a landscape: for a system with two degrees of freedom (e.g. two bond lengths), the value of the energy (analogy: the height of the land) is a function of two bond lengths (analogy: the coordinates of the position on the ground).
The PES concept finds application in fields such as physics, chemistry and biochemistry, especially in the theoretical sub-branches of these subjects. It can be used to theoretically explore properties of structures composed of atoms, for example, finding the minimum energy shape of a molecule or computing the rates of a chemical reaction. It can be used to describe all possible conformations of a molecular entity, or the spatial positions of interacting molecules in a system, or parameters and their corresponding energy levels, typically Gibbs free energy. Geometrically, the energy landscape is the graph of the energy function across the configuration space of the system. The term is also used more generally in geometric perspectives to mathematical optimization, when the domain of the loss function is the parameter space of some system.
The geometry of a set of atoms can be described by a vector, r, whose elements represent the atom positions. The vector r could be the set of the Cartesian coordinates of the atoms, or could also be a set of inter-atomic distances and angles.
Given r, the energy as a function of the positions, E(r), is the value of E(r) for all r of interest. Using the landscape analogy from the introduction, E gives the height on the "energy landscape" so that the concept of a potential energy surface arises.
To study a chemical reaction using the PES as a function of atomic positions, it is necessary to calculate the energy for every atomic arrangement of interest. Methods of calculating the energy of a particular atomic arrangement of atoms are well described in the computational chemistry article, and the emphasis here will be on finding approximations of E(r) to yield fine-grained energy-position information.
For very simple chemical systems or when simplifying approximations are made about inter-atomic interactions, it is sometimes possible to use an analytically derived expression for the energy as a function of the atomic positions. An example is the London-Eyring-Polanyi-Sato potential for the system H + H2 as a function of the three H-H distances.
For more complicated systems, calculation of the energy of a particular arrangement of atoms is often too computationally expensive for large scale representations of the surface to be feasible. For these systems a possible approach is to calculate only a reduced set of points on the PES and then use a computationally cheaper interpolation method, for example Shepard interpolation, to fill in the gaps.
Hub AI
Potential energy surface AI simulator
(@Potential energy surface_simulator)
Potential energy surface
A potential energy surface (PES) or energy landscape describes the energy of a system, especially a collection of atoms, in terms of certain parameters, normally the positions of the atoms. The surface might define the energy as a function of one or more coordinates; if there is only one coordinate, the surface is called a potential energy curve or energy profile. An example is the Morse/Long-range potential.
It is helpful to use the analogy of a landscape: for a system with two degrees of freedom (e.g. two bond lengths), the value of the energy (analogy: the height of the land) is a function of two bond lengths (analogy: the coordinates of the position on the ground).
The PES concept finds application in fields such as physics, chemistry and biochemistry, especially in the theoretical sub-branches of these subjects. It can be used to theoretically explore properties of structures composed of atoms, for example, finding the minimum energy shape of a molecule or computing the rates of a chemical reaction. It can be used to describe all possible conformations of a molecular entity, or the spatial positions of interacting molecules in a system, or parameters and their corresponding energy levels, typically Gibbs free energy. Geometrically, the energy landscape is the graph of the energy function across the configuration space of the system. The term is also used more generally in geometric perspectives to mathematical optimization, when the domain of the loss function is the parameter space of some system.
The geometry of a set of atoms can be described by a vector, r, whose elements represent the atom positions. The vector r could be the set of the Cartesian coordinates of the atoms, or could also be a set of inter-atomic distances and angles.
Given r, the energy as a function of the positions, E(r), is the value of E(r) for all r of interest. Using the landscape analogy from the introduction, E gives the height on the "energy landscape" so that the concept of a potential energy surface arises.
To study a chemical reaction using the PES as a function of atomic positions, it is necessary to calculate the energy for every atomic arrangement of interest. Methods of calculating the energy of a particular atomic arrangement of atoms are well described in the computational chemistry article, and the emphasis here will be on finding approximations of E(r) to yield fine-grained energy-position information.
For very simple chemical systems or when simplifying approximations are made about inter-atomic interactions, it is sometimes possible to use an analytically derived expression for the energy as a function of the atomic positions. An example is the London-Eyring-Polanyi-Sato potential for the system H + H2 as a function of the three H-H distances.
For more complicated systems, calculation of the energy of a particular arrangement of atoms is often too computationally expensive for large scale representations of the surface to be feasible. For these systems a possible approach is to calculate only a reduced set of points on the PES and then use a computationally cheaper interpolation method, for example Shepard interpolation, to fill in the gaps.