Community hub
Recent from talks
Knowledge base stats:
Talk channels stats:
Members stats:
MYD88
Myeloid differentiation primary response 88 (MYD88) is a protein that, in humans, is encoded by the MYD88 gene. originally discovered in the laboratory of Dan A. Liebermann (Lord et al. Oncogene 1990) as a Myeloid differentiation primary response gene.
The MYD88 gene provides instructions for making a protein involved in signaling within immune cells. The MyD88 protein acts as an adapter, connecting proteins that receive signals from outside the cell to the proteins that relay signals inside the cell.
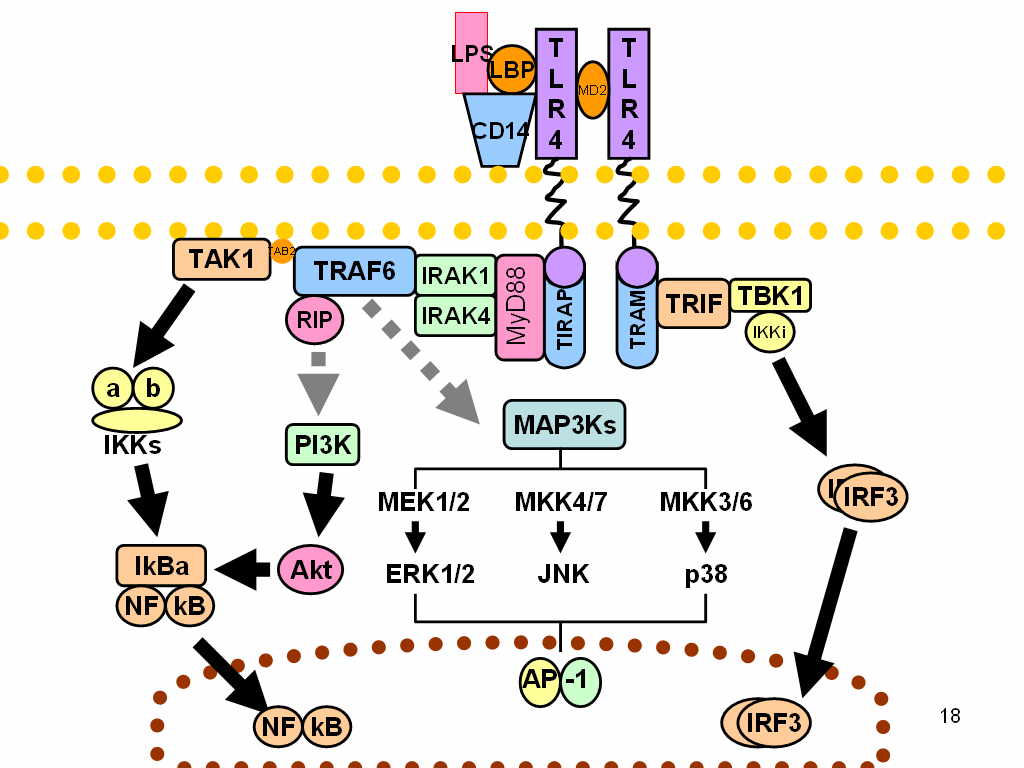
In innate immunity, the MyD88 plays a pivotal role in immune cell activation through Toll-like receptors (TLRs), which belong to large group of pattern recognition receptors (PRR). In general, these receptors sense common patterns which are shared by various pathogens – Pathogen-associated molecular pattern (PAMPs), or which are produced/released during cellular damage – damage-associated molecular patterns (DAMPs).
TLRs are homologous to Toll receptors, which were first described in the ontogenesis of fruit flies Drosophila, being responsible for dorso-ventral development. Hence, TLRs have been proved in all animals from insects to mammals. TLRs are located either on the cellular surface (TLR1, TLR2, TLR4, TLR5, TLR6) or within endosomes (TLR3, TLR7, TLR8, TLR9) sensing extracellular or phagocytosed pathogens, respectively. TLRs are integral membrane glycoproteins with typical semicircular-shaped extracellular parts containing leucine-rich repeats responsible for ligand binding, and Intracellular parts containing Toll-Interleukin receptor (TIR) domain.
After ligand binding, all TLRs, apart from TLR3, interact with adaptor protein MyD88. Another adaptor protein, which is activated by TLR3 and TLR4, is called TIR domain-containing adapter-inducing IFN-β (TRIF). Subsequently, these proteins activate two important transcription factors:
TLR7 and TLR9 activate both NF-κB and IRF3 through MyD88-dependent and TRIF-independent pathway, respectively.
The human ortholog MYD88 seems to function similarly to mice, since the immunological phenotype of human cells deficient in MYD88 is similar to cells from MyD88 deficient mice. However, available evidence suggests that MYD88 is dispensable for human resistance to common viral infections and to all but a few pyogenic bacterial infections, demonstrating a major difference between mouse and human immune responses. Mutation in MYD88 at position 265 leading to a change from leucine to proline have been identified in many human lymphomas including ABC subtype of diffuse large B-cell lymphoma and Waldenström's macroglobulinemia.
Myd88 has been shown to interact with:
Hub AI
MYD88 AI simulator
(@MYD88_simulator)
MYD88
Myeloid differentiation primary response 88 (MYD88) is a protein that, in humans, is encoded by the MYD88 gene. originally discovered in the laboratory of Dan A. Liebermann (Lord et al. Oncogene 1990) as a Myeloid differentiation primary response gene.
The MYD88 gene provides instructions for making a protein involved in signaling within immune cells. The MyD88 protein acts as an adapter, connecting proteins that receive signals from outside the cell to the proteins that relay signals inside the cell.
In innate immunity, the MyD88 plays a pivotal role in immune cell activation through Toll-like receptors (TLRs), which belong to large group of pattern recognition receptors (PRR). In general, these receptors sense common patterns which are shared by various pathogens – Pathogen-associated molecular pattern (PAMPs), or which are produced/released during cellular damage – damage-associated molecular patterns (DAMPs).
TLRs are homologous to Toll receptors, which were first described in the ontogenesis of fruit flies Drosophila, being responsible for dorso-ventral development. Hence, TLRs have been proved in all animals from insects to mammals. TLRs are located either on the cellular surface (TLR1, TLR2, TLR4, TLR5, TLR6) or within endosomes (TLR3, TLR7, TLR8, TLR9) sensing extracellular or phagocytosed pathogens, respectively. TLRs are integral membrane glycoproteins with typical semicircular-shaped extracellular parts containing leucine-rich repeats responsible for ligand binding, and Intracellular parts containing Toll-Interleukin receptor (TIR) domain.
After ligand binding, all TLRs, apart from TLR3, interact with adaptor protein MyD88. Another adaptor protein, which is activated by TLR3 and TLR4, is called TIR domain-containing adapter-inducing IFN-β (TRIF). Subsequently, these proteins activate two important transcription factors:
TLR7 and TLR9 activate both NF-κB and IRF3 through MyD88-dependent and TRIF-independent pathway, respectively.
The human ortholog MYD88 seems to function similarly to mice, since the immunological phenotype of human cells deficient in MYD88 is similar to cells from MyD88 deficient mice. However, available evidence suggests that MYD88 is dispensable for human resistance to common viral infections and to all but a few pyogenic bacterial infections, demonstrating a major difference between mouse and human immune responses. Mutation in MYD88 at position 265 leading to a change from leucine to proline have been identified in many human lymphomas including ABC subtype of diffuse large B-cell lymphoma and Waldenström's macroglobulinemia.
Myd88 has been shown to interact with: