Community hub
Pyoderma gangrenosum
View on Wikipedia| Pyoderma gangrenosum | |
|---|---|
 | |
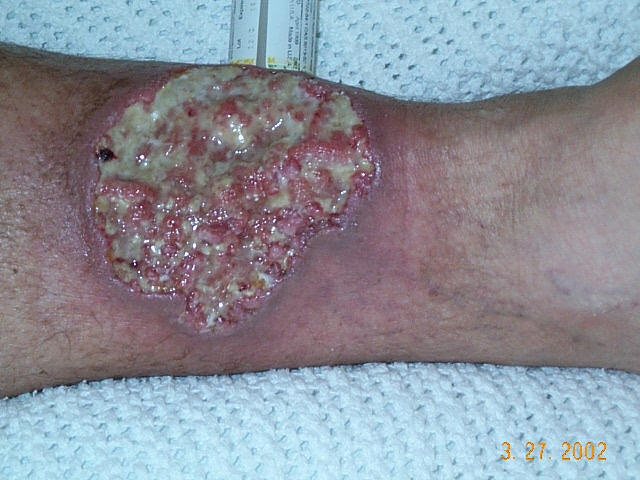
| Pyoderma gangrenosum on the leg of a person with ulcerative colitis. | |
| Specialty | Dermatology |
| Usual onset | 40s or 50s[1] |
| Treatment | Corticosteroids, ciclosporin, infliximab, canakinumab[2] |
Pyoderma gangrenosum is a rare, inflammatory skin disease where painful pustules or nodules become ulcers that progressively grow.[3] Pyoderma gangrenosum is not infectious.[3]
Treatments may include corticosteroids, ciclosporin, infliximab, or canakinumab.[2]
The disease was identified in 1908. It affects approximately 1 person in 100,000 in the population. Though it can affect people of any age, it mostly affects people in their 40s and 50s.[1]
Types
[edit]
There are two main types of pyoderma gangrenosum:[1]
- the 'typical' ulcerative form, which occurs in the legs
- an 'atypical' form that is more superficial and occurs in the hands and other parts of the body
Other variations are:[4]
- Peristomal pyoderma gangrenosum comprises 15% of all cases of pyoderma
- Bullous pyoderma gangrenosum
- Pustular pyoderma gangrenosum[5]
- Vegetative pyoderma gangrenosum[6]
Presentation
[edit]Associations
[edit]The following are conditions commonly associated with pyoderma gangrenosum:[7][8]
- Inflammatory bowel disease:
- Arthritides:
- Hematological disease:
- Solid tumors
A rare[10] syndromic association called pyogenic arthritis, pyoderma gangrenosum and acne syndrome (PAPA syndrome), a type of autoinflammatory disorder, is associated with mutations in the proline-serine-threonine phosphatase-interacting 1 gene (PSTPIP1).[10][11]
Causes
[edit]Though the cause is not well understood, the disease is thought to be due to immune system dysfunction, and particularly improper functioning of neutrophils. In support of an immune cause, a variety of immune mediators such as interleukin (IL)-8, IL-1β, IL-6, interferon (IFN)-γ, granulocyte colony-stimulating factor, tumor necrosis factor alpha, matrix metalloproteinase (MMP)-9, MMP10, and elafin have all been reported to be elevated in patients with pyoderma gangrenosum.[12]
Also in support of an immune cause is the finding that at least half of all pyoderma gangrenosum patients suffer from immune-mediated diseases.[1] For instance, ulcerative colitis, rheumatoid arthritis,[4] and monoclonal gammopathies[13] have all been associated with pyoderma gangrenosum. It can also be part of autoinflammatory syndromes such as PAPA syndrome.[10][11] Marzano et al. (2017) identified a variety of single-nucleotide polymorphisms (SNPs) linked to autoinflammation that were carried, singly or in combination, in subsets of patients with pyoderma gangrenosum, acne and suppurative hidradenitis syndrome (PASH syndrome) or isolated pyoderma gangrenosum of the ulcerative subtype.[14]
One hallmark of pyoderma gangrenosum is pathergy, which is the appearance of new lesions at sites of trauma.[15]
Diagnosis
[edit]Diagnosis of PG is challenging owing to its variable presentation, clinical overlap with other conditions, association with several systemic diseases, and absence of defining histopathologic or laboratory findings. Misdiagnosis and delayed diagnosis are common. It has been shown that up to 39% of patients who initially received a diagnosis of PG have an alternative diagnosis.[16] In light of this, validated diagnostic criteria have recently been developed for ulcerative pyoderma gangrenosum.[17]
Diagnostic criteria
[edit]In addition to a biopsy demonstrating a neutrophilic infiltrate, patients must have at least 4 minor criteria to meet diagnostic criteria.[17] These criteria are based on histology, history, clinical examination, and treatment.[citation needed]
- Histology: Exclusion of infection (including histologically indicated stains and tissue cultures)
- Pathergy (ulcer occurring at sites of trauma, with the ulcer extending past the area of trauma)
- Personal history of inflammatory bowel disease or inflammatory arthritis
- History of papule, pustule, or vesicle that rapidly ulcerated
- Clinical examination (or photographic evidence) of peripheral erythema, undermining border, and tenderness at the site of ulceration
- Multiple ulcerations (at least 1 occurring on an anterior lower leg)
- Cribriform or “wrinkled paper” scars at sites of healed ulcers
- Decrease in ulcer size within 1 month of initiating immunosuppressive medications
Treatment
[edit]First-line therapy for disseminated or localized instances of pyoderma gangrenosum is systemic treatment with corticosteroids and ciclosporin. Topical application of clobetasol, mupirocin, and gentamicin alternated with tacrolimus can be effective. Pyoderma gangrenosum ulcers demonstrate pathergy, that is, a worsening in response to minor trauma or surgical debridement. Significant care should be taken with dressing changes to prevent potentially rapid wound growth. Many patients respond differently to different types of treatment, for example, some benefit from a moist environment, so treatment should be carefully evaluated at each stage.[citation needed]
If ineffective, alternative therapeutic procedures include systemic treatment with corticosteroids and mycophenolate mofetil; mycophenolate mofetil and ciclosporin; tacrolimus; thalidomide; infliximab; or plasmapheresis.[18]
See also
[edit]References
[edit]- ^ a b c d Jackson JM, Callen JP (April 23, 2012). Elston DM (ed.). "Pyoderma Gangrenosum". EMedicine.
- ^ a b Partridge AC, Bai JW, Rosen CF, Walsh SR, Gulliver WP, Fleming P (August 2018). "Effectiveness of systemic treatments for pyoderma gangrenosum: a systematic review of observational studies and clinical trials". The British Journal of Dermatology. 179 (2): 290–295. doi:10.1111/bjd.16485. PMID 29478243. S2CID 3504429.
- ^ a b Ruocco E, Sangiuliano S, Gravina AG, Miranda A, Nicoletti G (September 2009). "Pyoderma gangrenosum: an updated review". Journal of the European Academy of Dermatology and Venereology. 23 (9): 1008–17. doi:10.1111/j.1468-3083.2009.03199.x. PMID 19470075. S2CID 29773727.
- ^ a b Brooklyn T, Dunnill G, Probert C (July 2006). "Diagnosis and treatment of pyoderma gangrenosum". BMJ. 333 (7560): 181–4. doi:10.1136/bmj.333.7560.181. PMC 1513476. PMID 16858047.
- ^ Shankar S, Sterling JC, Rytina E (November 2003). "Pustular pyoderma gangrenosum". Clinical and Experimental Dermatology. 28 (6): 600–3. doi:10.1046/j.1365-2230.2003.01418.x. PMID 14616824. S2CID 11350602.
- ^ Langan SM, Powell FC (August 2005). "Vegetative pyoderma gangrenosum: a report of two new cases and a review of the literature". International Journal of Dermatology. 44 (8): 623–9. doi:10.1111/j.1365-4632.2005.02591.x. PMID 16101860. S2CID 34574262.
- ^ Brooklyn T, Dunnill G, Probert C (July 2006). "Diagnosis and treatment of pyoderma gangrenosum". BMJ. 333 (7560): 181–4. doi:10.1136/bmj.333.7560.181. PMC 1513476. PMID 16858047.
- ^ Schmieder SJ, Krishnamurthy K (4 July 2023). "Pyoderma Gangrenosum". StatPearls. Treasure Island, Florida: StatPearls Publishing. PMID 29489279.
- ^ Tendas A, Niscola P, Barbati R, Abruzzese E, Cuppelli L, Giovannini M, et al. (May 2011). "Tattoo related pyoderma/ectyma gangrenous as presenting feature of relapsed acute myeloid leukaemia: an exceptionally rare observation". Injury. 42 (5): 546–7. doi:10.1016/j.injury.2010.08.014. PMID 20883993.
- ^ a b c Genovese G, Moltasio C, Garcovich S, Marzano AV (2020). "PAPA Spectrum Disorders". Italian Journal of Dermatology and Venereology. 155 (5): 542–550. doi:10.23736/S0392-0488.20.06629-8. PMID 32618443.
- ^ a b Smith EJ, Allantaz F, Bennett L, Zhang D, Gao X, Wood G, Kastner DL, Punaro M, Aksentijevich I, Pascual V, Wise CA (2010). "Clinical, Molecular, and Genetic Characteristics of PAPA Syndrome: A Review". Current Genomics. 11 (7): 519–527. doi:10.2174/138920210793175921. PMC 3048314. PMID 21532836.
- ^ Patel F, Fitzmaurice S, Duong C, He Y, Fergus J, Raychaudhuri SP, et al. (May 2015). "Effective strategies for the management of pyoderma gangrenosum: a comprehensive review". Acta Dermato-Venereologica. 95 (5): 525–31. doi:10.2340/00015555-2008. PMID 25387526.
- ^ Claveau JS, Wetter DA, Kumar S (2022). "Cutaneous manifestations of monoclonal gammopathy". Blood Cancer Journal. 12 (4). doi:10.1038/s41408-022-00661-1. PMC 9001632. PMID 35411042. 58.
- ^ Marzano AV, Damiani G, Ceccherini I, Berti E, Gattorno M, Cugno M (2017). "Autoinflammation in pyoderma gangrenosum and its syndromic form (pyoderma gangrenosum, acne and suppurative hidradenitis)". British Journal of Dermatology. 176 (6): 1588–1598. doi:10.1111/bjd.15226. PMID 27943240.
- ^ Rashid RM (November 2008). "Seat belt pyoderma gangrenosum: minor pressure as a causative factor". Journal of the European Academy of Dermatology and Venereology. 22 (10): 1273–4. doi:10.1111/j.1468-3083.2008.02626.x. PMID 18837131. S2CID 27476857.
- ^ Weenig RH, Davis MD, Dahl PR, Su WP (October 2002). "Skin ulcers misdiagnosed as pyoderma gangrenosum". The New England Journal of Medicine. 347 (18): 1412–8. doi:10.1056/NEJMoa013383. PMID 12409543.
- ^ a b Maverakis E, Ma C, Shinkai K, Fiorentino D, Callen JP, Wollina U, et al. (April 2018). "Diagnostic Criteria of Ulcerative Pyoderma Gangrenosum: A Delphi Consensus of International Experts". JAMA Dermatology. 154 (4): 461–466. doi:10.1001/jamadermatol.2017.5980. hdl:2434/555642. PMID 29450466. S2CID 4774649.
- ^ Reichrath J, Bens G, Bonowitz A, Tilgen W (August 2005). "Treatment recommendations for pyoderma gangrenosum: an evidence-based review of the literature based on more than 350 patients". Journal of the American Academy of Dermatology. 53 (2): 273–83. doi:10.1016/j.jaad.2004.10.006. PMID 16021123.