
Community hub
0 subscribers8 pages, 0 posts
Recent from talks
All channels
Be the first to start a discussion here.
Be the first to start a discussion here.
Be the first to start a discussion here.
Be the first to start a discussion here.
Contribute something
Welcome to the community hub built to collect knowledge and have discussions related to Keratoderma.
Nothing was collected or created yet.
Keratoderma
View on Wikipediafrom Wikipedia
| Keratoderma | |
|---|---|
 | |
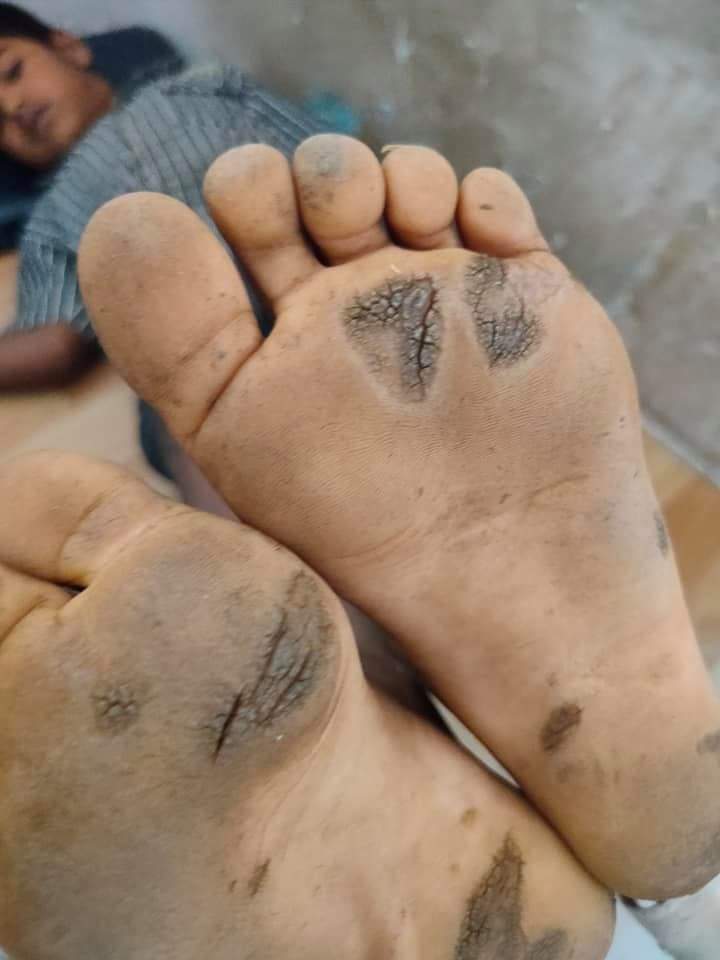
| Diffuse keratodermas affect most of the palms and soles. | |
| Specialty | Dermatology |
Keratoderma is a local or general thickening of the horny layer of the epidermis.[1]
Classification
[edit]The keratodermas are classified into the following subgroups:[2]: 506
Congenital
[edit]- Simple keratodermas
- Complex keratodermas
- Diffuse palmoplantar keratoderma
- Focal palmoplantar keratoderma
- Ectodermal dysplasias
- Syndromic keratodermas
- Vohwinkel syndrome
- Palmoplantar keratoderma associated with esophageal cancer
- Palmoplantar keratoderma and spastic paraplegia
- Naxos disease
- Striate palmoplantar keratoderma, woolly hair, and left ventricular dilated cardiomyopathy
- Keratitis-ichthyosis-deafness syndrome
- Corneodermatosseous syndrome
- Huriez syndrome
- Oculocutaneous tyrosinemia
- Cardiofaciocutaneous syndrome
- Schöpf–Schulz–Passarge syndrome
Acquired
[edit]- Acquired keratodermas
- AIDS-associated keratoderma
- Arsenical keratoses
- Calluses
- Climacteric keratoderma
- Clavi (Corns)
- Eczema
- Human papillomavirus
- Keratoderma blenorrhagicum
- Lichen planus
- Norwegian scabies
- Paraneoplastic keratoderma
- Psoriasis
- Reactive arthritis
- Secondary syphilis
- Tinea pedis
- Sézary syndrome
- Tuberculosis verrucosa cutis
- Drug-induced keratoderma[3]
See also
[edit]References
[edit]- ^ "keratoderma". Oxford English Dictionary (Online ed.). Oxford University Press. (Subscription or participating institution membership required.)
- ^ Freedberg, et al. (2003). Fitzpatrick's Dermatology in General Medicine. (6th ed.). McGraw-Hill. ISBN 0-07-138076-0.
- ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. p. 778. ISBN 978-1-4160-2999-1.
External links
[edit]This cutaneous condition article is a stub. You can help Wikipedia by expanding it. |
Keratoderma
View on Grokipediafrom Grokipedia