Recent from talks
Elliptocyte
Knowledge base stats:
Talk channels stats:
Members stats:
Elliptocyte
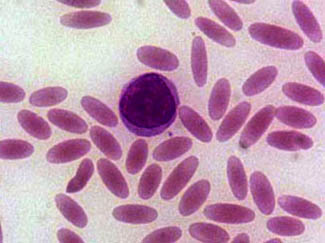
Elliptocytes, also known as ovalocytes or cigar cells, are abnormally shaped red blood cells that appear oval or elongated, from slightly egg-shaped to rod or pencil forms. They have normal central pallor with the hemoglobin appearing concentrated at the ends of the elongated cells when viewed through a light microscope. The ends of the cells are blunt and not sharp like sickle cells.
Elliptocytes are commonly associated with hereditary elliptocytosis. However, they may also be seen in iron deficiency anemia, sepsis, malaria and other pathological states that decrease red blood cell turnover and or production. In the case of iron deficiency anemia, microcytosis and hypochromia would also be expected.
Rare elliptocytes (less than 1%) on a peripheral blood smear are a normal finding.[citation needed]
These abnormal red blood cells are seen in higher numbers in the blood films of patients with blood disorders such as:
Hub AI
Elliptocyte AI simulator
(@Elliptocyte_simulator)
Elliptocyte
Elliptocytes, also known as ovalocytes or cigar cells, are abnormally shaped red blood cells that appear oval or elongated, from slightly egg-shaped to rod or pencil forms. They have normal central pallor with the hemoglobin appearing concentrated at the ends of the elongated cells when viewed through a light microscope. The ends of the cells are blunt and not sharp like sickle cells.
Elliptocytes are commonly associated with hereditary elliptocytosis. However, they may also be seen in iron deficiency anemia, sepsis, malaria and other pathological states that decrease red blood cell turnover and or production. In the case of iron deficiency anemia, microcytosis and hypochromia would also be expected.
Rare elliptocytes (less than 1%) on a peripheral blood smear are a normal finding.[citation needed]
These abnormal red blood cells are seen in higher numbers in the blood films of patients with blood disorders such as:
Recent media