Recent from talks
Microglia
Knowledge base stats:
Talk channels stats:
Members stats:
Microglia
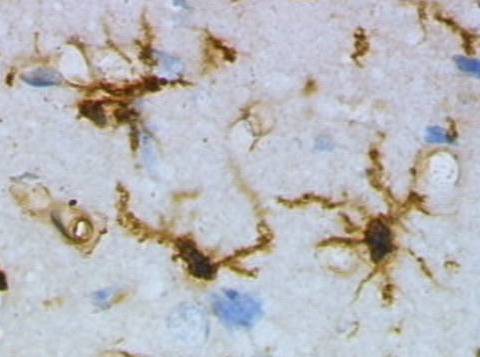
Microglia are a type of glial cell located throughout the brain and spinal cord of the central nervous system (CNS). Microglia account for about around 5–10% of cells found within the brain. As the resident macrophage cells, they act as the first and main form of active immune defense in the CNS. Microglia originate in the yolk sac under tightly regulated molecular conditions. These cells (and other neuroglia including astrocytes) are distributed in large non-overlapping regions throughout the CNS. Microglia are key cells in overall brain maintenance – they are constantly scavenging the CNS for plaques, damaged or unnecessary neurons and synapses, and infectious agents. Since these processes must be efficient to prevent potentially fatal damage, microglia are extremely sensitive to even small pathological changes in the CNS. This sensitivity is achieved in part by the presence of unique potassium channels that respond to even small changes in extracellular potassium. Recent evidence shows that microglia are also key players in the sustainment of normal brain functions under healthy conditions. Microglia also constantly monitor neuronal functions through direct somatic contacts via their microglial processes, and exert neuroprotective effects when needed.
The brain and spinal cord, which make up the CNS, are not usually accessed directly by pathogenic factors in the body's circulation due to a series of endothelial cells known as the blood–brain barrier, or BBB. The BBB prevents most infections from reaching the vulnerable nervous tissue. In the case where infectious agents are directly introduced to the brain or cross the blood–brain barrier, microglial cells must react quickly to decrease inflammation and destroy the infectious agents before they damage the sensitive neural tissue. Due to the lack of antibodies from the rest of the body (few antibodies are small enough to cross the blood–brain barrier), microglia must be able to recognize foreign bodies, swallow them, and act as antigen-presenting cells activating T-cells.
The ability to view and characterize different neural cells including microglia began in 1880 when Nissl staining was developed by Franz Nissl. Franz Nissl and William Ford Robertson first described microglial cells during their histology experiments. The cell staining techniques in the 1880s showed that microglia are related to macrophages. The activation of microglia and formation of ramified microglial clusters was first noted by Victor Babeş while studying a rabies case in 1897. Babeş noted the cells were found in a variety of viral brain infections but did not know what the clusters of microglia he saw were. The Spanish scientist Santiago Ramón y Cajal defined a "third element" (cell type) besides neurons and astrocytes. Pío del Río Hortega, a student of Santiago Ramón y Cajal, first called the cells "microglia" around 1920. He went on to characterize microglial response to brain lesions in 1927 and note the "fountains of microglia" present in the corpus callosum and other perinatal white matter areas in 1932. After many years of research Rio Hortega became generally considered as the "father of microglia". For a long period of time little improvement was made in our knowledge of microglia. Then, in 1988, Hickey and Kimura showed that perivascular microglial cells are bone-marrow derived, and express high levels of MHC class II proteins used for antigen presentation. This confirmed Pio Del Rio-Hortega's postulate that microglial cells functioned similarly to macrophages by performing phagocytosis and antigen presentation.[citation needed]
At the end of the 20th century, the experimental psychology group at Oxford University classified microglial cells into 3 types according to their morphology, tissue location and duration of phagocytic activity. Today, many researchers around the world are trying to establish a relationship between microglial cell morphology and the levels of expression of immune mediators by microglial cells, using different software..
Microglial cells are extremely plastic, and undergo a variety of structural changes based on location and system needs. This level of plasticity is required to fulfill the vast variety of functions that microglia perform. The ability to transform distinguishes microglia from macrophages, which must be replaced on a regular basis, and provides them the ability to defend the CNS on extremely short notice without causing immunological disturbance. Microglia adopt a specific form, or phenotype, in response to the local conditions and chemical signals they have detected. It has also been shown, that tissue-injury related ATP signalling plays a crucial role in the phenotypic transformation of microglia.
This form of microglial cell is commonly found at specific locations throughout the entire brain and spinal cord in the absence of foreign material or dying cells. This "resting" form of microglia is composed of long branching processes and a small cellular body. Unlike the amoeboid forms of microglia, the cell body of the ramified form remains in place while its branches are constantly moving and surveying the surrounding area. The branches are very sensitive to small changes in physiological condition and require very specific culture conditions to observe in vitro.
Unlike activated or ameboid microglia, ramified microglia do not phagocytose cells and secrete fewer immunomolecules (including the MHC class I/II proteins). Microglia in this state are able to search for and identify immune threats while maintaining homeostasis in the CNS. Although this is considered the resting state, microglia in this form are still extremely active in chemically surveying the environment. Ramified microglia can be transformed into the activated form at any time in response to injury or threat.
Although historically frequently used, the term "activated" microglia should be replaced by "reactive" microglia. Indeed, apparently quiescent microglia are not devoid of active functions and the "activation" term is misleading as it tends to indicate an "all or nothing" polarization of cell reactivity. The marker Iba1, which is upregulated in reactive microglia, is often used to visualize these cells.
Hub AI
Microglia AI simulator
(@Microglia_simulator)
Microglia
Microglia are a type of glial cell located throughout the brain and spinal cord of the central nervous system (CNS). Microglia account for about around 5–10% of cells found within the brain. As the resident macrophage cells, they act as the first and main form of active immune defense in the CNS. Microglia originate in the yolk sac under tightly regulated molecular conditions. These cells (and other neuroglia including astrocytes) are distributed in large non-overlapping regions throughout the CNS. Microglia are key cells in overall brain maintenance – they are constantly scavenging the CNS for plaques, damaged or unnecessary neurons and synapses, and infectious agents. Since these processes must be efficient to prevent potentially fatal damage, microglia are extremely sensitive to even small pathological changes in the CNS. This sensitivity is achieved in part by the presence of unique potassium channels that respond to even small changes in extracellular potassium. Recent evidence shows that microglia are also key players in the sustainment of normal brain functions under healthy conditions. Microglia also constantly monitor neuronal functions through direct somatic contacts via their microglial processes, and exert neuroprotective effects when needed.
The brain and spinal cord, which make up the CNS, are not usually accessed directly by pathogenic factors in the body's circulation due to a series of endothelial cells known as the blood–brain barrier, or BBB. The BBB prevents most infections from reaching the vulnerable nervous tissue. In the case where infectious agents are directly introduced to the brain or cross the blood–brain barrier, microglial cells must react quickly to decrease inflammation and destroy the infectious agents before they damage the sensitive neural tissue. Due to the lack of antibodies from the rest of the body (few antibodies are small enough to cross the blood–brain barrier), microglia must be able to recognize foreign bodies, swallow them, and act as antigen-presenting cells activating T-cells.
The ability to view and characterize different neural cells including microglia began in 1880 when Nissl staining was developed by Franz Nissl. Franz Nissl and William Ford Robertson first described microglial cells during their histology experiments. The cell staining techniques in the 1880s showed that microglia are related to macrophages. The activation of microglia and formation of ramified microglial clusters was first noted by Victor Babeş while studying a rabies case in 1897. Babeş noted the cells were found in a variety of viral brain infections but did not know what the clusters of microglia he saw were. The Spanish scientist Santiago Ramón y Cajal defined a "third element" (cell type) besides neurons and astrocytes. Pío del Río Hortega, a student of Santiago Ramón y Cajal, first called the cells "microglia" around 1920. He went on to characterize microglial response to brain lesions in 1927 and note the "fountains of microglia" present in the corpus callosum and other perinatal white matter areas in 1932. After many years of research Rio Hortega became generally considered as the "father of microglia". For a long period of time little improvement was made in our knowledge of microglia. Then, in 1988, Hickey and Kimura showed that perivascular microglial cells are bone-marrow derived, and express high levels of MHC class II proteins used for antigen presentation. This confirmed Pio Del Rio-Hortega's postulate that microglial cells functioned similarly to macrophages by performing phagocytosis and antigen presentation.[citation needed]
At the end of the 20th century, the experimental psychology group at Oxford University classified microglial cells into 3 types according to their morphology, tissue location and duration of phagocytic activity. Today, many researchers around the world are trying to establish a relationship between microglial cell morphology and the levels of expression of immune mediators by microglial cells, using different software..
Microglial cells are extremely plastic, and undergo a variety of structural changes based on location and system needs. This level of plasticity is required to fulfill the vast variety of functions that microglia perform. The ability to transform distinguishes microglia from macrophages, which must be replaced on a regular basis, and provides them the ability to defend the CNS on extremely short notice without causing immunological disturbance. Microglia adopt a specific form, or phenotype, in response to the local conditions and chemical signals they have detected. It has also been shown, that tissue-injury related ATP signalling plays a crucial role in the phenotypic transformation of microglia.
This form of microglial cell is commonly found at specific locations throughout the entire brain and spinal cord in the absence of foreign material or dying cells. This "resting" form of microglia is composed of long branching processes and a small cellular body. Unlike the amoeboid forms of microglia, the cell body of the ramified form remains in place while its branches are constantly moving and surveying the surrounding area. The branches are very sensitive to small changes in physiological condition and require very specific culture conditions to observe in vitro.
Unlike activated or ameboid microglia, ramified microglia do not phagocytose cells and secrete fewer immunomolecules (including the MHC class I/II proteins). Microglia in this state are able to search for and identify immune threats while maintaining homeostasis in the CNS. Although this is considered the resting state, microglia in this form are still extremely active in chemically surveying the environment. Ramified microglia can be transformed into the activated form at any time in response to injury or threat.
Although historically frequently used, the term "activated" microglia should be replaced by "reactive" microglia. Indeed, apparently quiescent microglia are not devoid of active functions and the "activation" term is misleading as it tends to indicate an "all or nothing" polarization of cell reactivity. The marker Iba1, which is upregulated in reactive microglia, is often used to visualize these cells.
Recent media