
Community hub
Recent from talks
Contribute something
Nothing was collected or created yet.
End-plate potential
View on Wikipedia
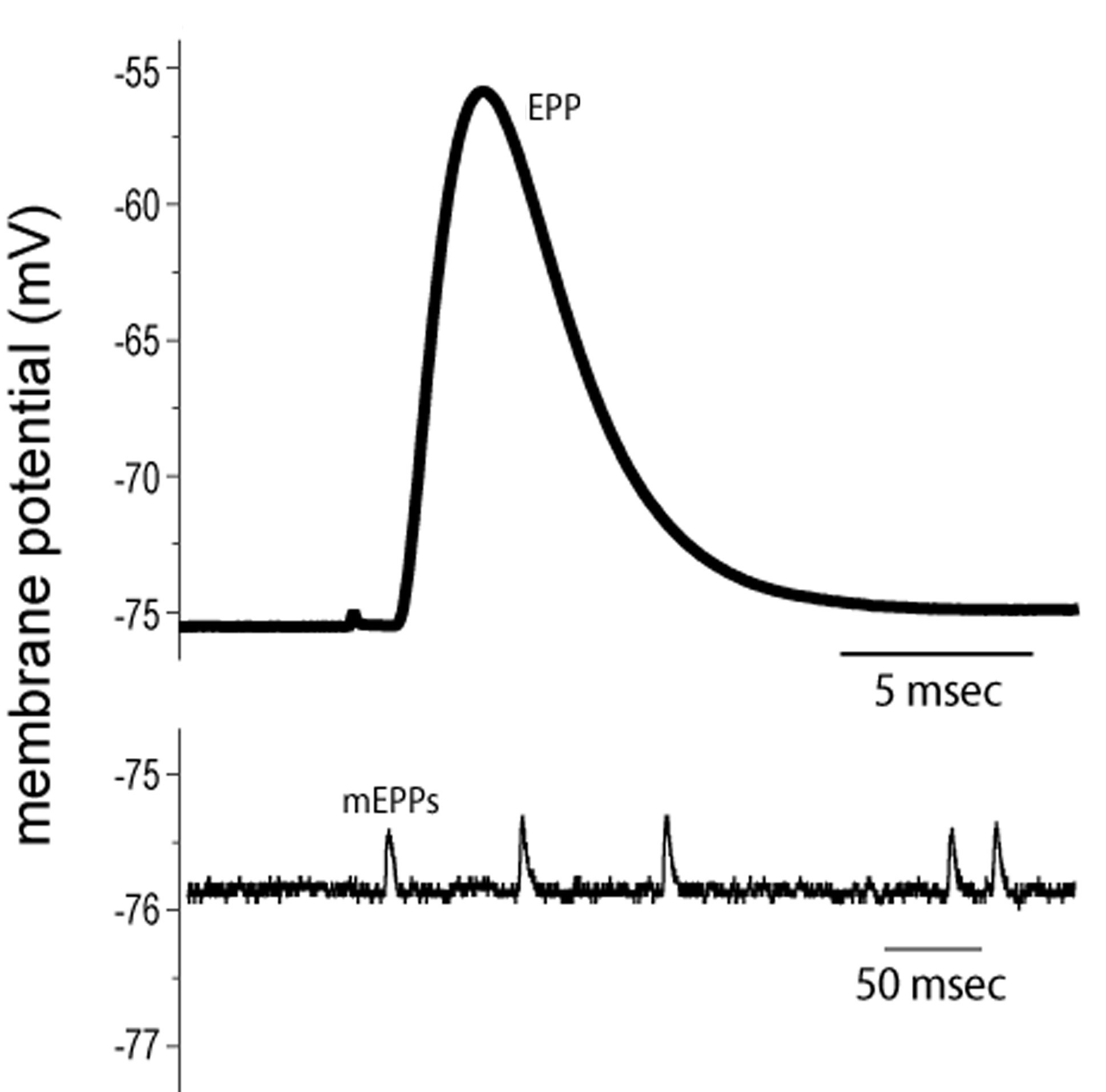
End plate potentials (EPPs) are the voltages which cause depolarization of skeletal muscle fibers caused by neurotransmitters binding to the postsynaptic membrane in the neuromuscular junction. They are called "end plates" because the postsynaptic terminals of muscle fibers have a large, saucer-like appearance. When an action potential reaches the axon terminal of a motor neuron, vesicles carrying neurotransmitters (mostly acetylcholine) are exocytosed and the contents are released into the neuromuscular junction. These neurotransmitters bind to receptors on the postsynaptic membrane and lead to its depolarization. In the absence of an action potential, acetylcholine vesicles spontaneously leak into the neuromuscular junction and cause very small depolarizations in the postsynaptic membrane. This small response (~0.4mV)[1] is called a miniature end plate potential (MEPP) and is generated by one acetylcholine-containing vesicle. It represents the smallest possible depolarization which can be induced in a muscle.
Neuromuscular junction
[edit]
The neuromuscular junction is the synapse that is formed between an alpha motor neuron (α-MN) and the skeletal muscle fiber. In order for a muscle to contract, an action potential is first propagated down a nerve until it reaches the axon terminal of the motor neuron. The motor neuron then innervates the muscle fibers to contraction by causing an action potential on the postsynaptic membrane of the neuromuscular junction.
Acetylcholine
[edit]End plate potentials are produced almost entirely by the neurotransmitter acetylcholine in skeletal muscle. Acetylcholine is the second most important excitatory neurotransmitter in the body following glutamate. It controls the somatosensory system which includes the senses of touch, vision, and hearing. It was the first neurotransmitter to be identified in 1914 by Henry Dale. Acetylcholine is synthesized in the cytoplasm of the neuron from choline and acetyl-CoA. Choline acetyltransferase is the enzyme that synthesizes acetylcholine and is often used as a marker in research relating to acetylcholine production. Neurons that utilize acetylcholine are called cholinergic neurons and they are very important in muscle contraction, memory, and learning.[2]
Ion channels
[edit]The polarization of membranes is controlled by sodium, potassium, calcium, and chloride ion channels. There are two types of ion channels involved in the neuromuscular junction and end plate potentials: voltage-gated ion channel and ligand-gated ion channel. Voltage gated ion channels are responsive to changes in membrane voltage which cause the voltage gated ion channel to open and allows certain ions to pass through. Ligand gated ion channels are responsive to certain molecules such as neurotransmitters. The binding of a ligand to the receptor on the ion channel protein causes a conformational change which allows the passing of certain ions.
Presynaptic membrane
[edit]Normally the resting membrane potential of a motor neuron is kept at -70mV to -50 with a higher concentration of sodium outside and a higher concentration of potassium inside. When an action potential propagates down a nerve and reaches the axon terminal of the motor neuron, the change in membrane voltage causes the calcium voltage gated ion channels to open allowing for an influx of calcium ions. These calcium ions cause the acetylcholine vesicles attached to the presynaptic membrane to release acetylcholine via exocytosis into the synaptic cleft.[3]
Postsynaptic membrane
[edit]EPP are caused mostly by the binding of acetylcholine to receptors in the postsynaptic membrane. There are two different kinds of acetylcholine receptors: nicotinic and muscarinic. Nicotinic receptors are ligand gated ion channels for fast transmission. All acetylcholine receptors in the neuromuscular junction are nicotinic. Muscarinic receptors are G protein-coupled receptors that use a second messenger. These receptors are slow and therefore are unable to measure a miniature end plate potential (MEPP). They are located in the parasympathetic nervous system such as in the vagus nerve and the gastrointestinal tract. During fetal development acetylcholine receptors are concentrated on the postsynaptic membrane and the entire surface of the nerve terminal in the growing embryo is covered even before a signal is fired. Five subunits consisting of four different proteins from four different genes comprise the nicotinic acetylcholine receptors therefore their packaging and assembly is a very complicated process with many different factors. The enzyme muscle-specific kinase (MuSK) initiates signaling processes in the developing postsynaptic muscle cell. It stabilizes the postsynaptic acetylcholine receptor clusters, facilitates the transcription of synaptic genes by muscle fiber nuclei, and triggers differentiation of the axon growth cone to form a differentiated nerve terminal.[4] Substrate laminin induces advanced maturation of the acetylcholine receptor clusters on the surfaces of myotubes.[5]
Initiation
[edit]Synaptic vesicles
[edit]All neurotransmitters are released into the synaptic cleft via exocytosis from synaptic vesicles. Two kinds of neurotransmitter vesicles exist: large dense core vesicles and small clear core vesicles. Large dense core vesicles contain neuropeptides and large neurotransmitters that are created in the cell body of the neuron and then transported via fast axonal transport down to the axon terminal. Small clear core vesicles transport small molecule neurotransmitters that are synthesized locally in the presynaptic terminals. Finalized neurotransmitter vesicles are bound to the presynaptic membrane. When an action potential propagates down the motor neuron axon and arrives at the axon terminal, it causes a depolarization of the axon terminal and opens calcium channels. This causes the release of the neurotransmitters via vesicle exocytosis.
After exocytosis, vesicles are recycled during a process known as the synaptic vesicle cycle. The retrieved vesicular membranes are passed through several intracellular compartments where they are modified to make new synaptic vesicles. They are then stored in a reserve pool until they are needed again for transport and release of neurotransmitters.
Unlike the reserve pool, the readily releasable pool of synaptic vesicles is ready to be activated. Vesicle depletion from the readily releasable pool occurs during high frequency stimulation of long duration and the size of the evoked EPP reduces. This neuromuscular depression is due to less neurotransmitter release during stimulation. In order for depletion not to occur, there must be a balance between repletion and depletion which can happen at low stimulation frequencies of less than 30 Hz.[6]
When a vesicle releases its neurotransmitters via exocytosis, it empties its entire contents into the synaptic cleft. Neurotransmitter release from vesicles is therefore stated to be quantal because only whole numbers of vesicles can be released. In 1970, Bernard Katz from the University of London won the Nobel Prize for Physiology or Medicine for statistically determining the quantal size of acetylcholine vesicles based on noise analysis in the neuromuscular junction. Using a book on mechanical statistics[clarification needed], he was able to infer the size of individual events going on at the same time.
The synaptic vesicles of acetylcholine are clear core synaptic vesicles with a diameter of 30 nm. Each acetylcholine vesicle contains approximately 5000 acetylcholine molecules. The vesicles release their entire quantity of acetylcholine and this causes miniature end plate potentials (MEPPs) to occur which are less than 1mV in amplitude and not enough to reach threshold.[7]
Miniature end plate potentials (MEPPs)
[edit]Miniature end plate potentials are the small (~0.4mV) depolarizations of the postsynaptic terminal caused by the release of a single vesicle into the synaptic cleft. Neurotransmitter vesicles containing acetylcholine collide spontaneously with the nerve terminal and release acetylcholine into the neuromuscular junction even without a signal from the axon. These small depolarizations are not enough to reach threshold and so an action potential in the postsynaptic membrane does not occur.[8] During experimentation with MEPPs, it was noticed that often spontaneous action potentials would occur, called end plate spikes in normal striated muscle without any stimulus. It was believed that these end plate spikes occurred as a result of injury or irritation of the muscles fibers due to the electrodes. Recent experiments have shown that these end plate spikes are actually caused by muscle spindles and have two distinct patterns: small and large. Small end plate spikes have a negative onset without signal propagation and large end plate spikes resemble motor unit potentials (MUPs). Muscle spindles are sensory receptors that measure muscle elongation or stretch and relay the information to the spinal cord or brain for the appropriate response.[9]
Threshold potential ("All or None")
[edit]When an action potential causes the release of many acetylcholine vesicles, acetylcholine diffuses across the neuromuscular junction and binds to ligand-gated nicotinic receptors (non-selective cation channels) on the muscle fiber. This allows for increased flow of sodium and potassium ions, causing depolarization of the sarcolemma (muscle cell membrane). The small depolarization associated with the release of acetylcholine from an individual synaptic vesicle is called a miniature end-plate potential (MEPP), and has a magnitude of about +0.4mV. MEPPs are additive, eventually increasing the end-plate potential (EPPs) from about -100mV up to the threshold potential of -60mV, at which level the voltage-gated ion channels in the postsynaptic membrane open, allowing a sudden flow of sodium ions from the synapse and a sharp spike in depolarization. This depolarization voltage spike triggers an action potential which propagates down the postsynaptic membrane leading to muscle contraction. It is important to note that EPPs are not action potentials, but that they trigger action potentials. In a normal muscular contraction, approximately 100-200 acetylcholine vesicles are released causing a depolarization that is 100 times greater in magnitude than a MEPP. This causes the membrane potential to depolarize +40mV (100 x 0.4mV = 40mV) from -100mV to -60mV where it reaches threshold.[7]
Action potential phases
[edit]Once the membrane potential reaches threshold, an action potential occurs and causes a sharp spike in membrane polarity. There are five phases of an action potential: threshold, depolarization, peak, repolarization, and hyperpolarization.
Threshold is when the summation of MEPPs reaches a certain potential and induces the opening of the voltage-gated ion channels. The rapid influx of sodium ions causes the membrane potential to reach a positive charge. The potassium ion channels are slower-acting than the sodium ion channels and so as the membrane potential starts to peak, the potassium ion channels open and causes an outflux of potassium to counteract the influx of sodium. At the peak, the outflux of potassium equals the influx of sodium, and the membrane does not change polarity.
During repolarization, the sodium channels begin to become inactivated, causing a net efflux of potassium ions. This causes the membrane potential to drop down to its resting membrane potential of -100mV. Hyperpolarization occurs because the slow-acting potassium channels take longer to deactivate, so the membrane overshoots the resting potential. It gradually returns to resting potential and is ready for another action potential to occur.
During the action potential before the hyperpolarization phase, the membrane is unresponsive to any stimulation. This inability to induce another action potential is known as the absolute refractory period. During the hyperpolarization period, the membrane is again responsive to stimulations but it requires a much higher input to induce an action potential. This phase is known as the relative refractory period.
Once the action potential has finished in the neuromuscular junction, the used acetylcholine is cleared out of the synaptic cleft by the enzyme acetylcholinesterase. Several diseases and problems can be caused by the inability of enzymes to clear away the neurotransmitters from the synaptic cleft leading to continued action potential propagation.[10]
Clinical applications
[edit]
Current research is attempting to learn more about end plate potentials and their effect on muscle activity. Many current diseases involve disrupted end plate potential activity. In Alzheimer patients, beta amyloid attaches to the acetylcholine receptors and inhibits acetylcholine binding. This causes less signal propagation and small EPPs that do not reach threshold. By analyzing brain processes with acetylcholine, doctors can measure how much beta amyloid is around and use it to judge its effects on Alzheimer's.[11] Myasthenia gravis is an autoimmune disease, where the body produces antibodies targeted against the acetylcholine receptor on the postsynaptic membrane in the neuromuscular junction. Muscle fatigue and weakness, worsened with use and improved by rest, is the hallmark of the disease. Because of the limited amount of acetylcholine receptors that are available for binding, symptomatic treatment consists of using an acetylcholinesterase inhibitor to reduce the breakdown of acetylcholine in the neuromuscular junction, so that enough acetylcholine will be present for the small number of unblocked receptors. A congenital abnormality caused by a deficiency in end-plate acetylcholine esterase (AChE) might be a pathophysiologic mechanism for myasthenic gravis. In a study on a patient with AChE deficiency, doctors noted that he had developed severe proximal and truncal muscle weakness with jittering in other muscles. It was found that a combination of the jitter and blocking rate of the acetylcholine receptors caused a reduced end-plate potential similar to what is seen in cases of myasthenia gravis.[12] Research of motor unit potentials (MUPs) has led to possible clinical applications in the evaluation of the progression of pathological diseases to myogenic or neurogenic origins by measuring the irregularity constant related. Motor unit potentials are the electrical signals produced by motor units that can be characterized by amplitude, duration, phase, and peak, and the irregularity coefficient (IR) is calculated based on the peak numbers and amplitudes.[13] Lambert–Eaton myasthenic syndrome is a disorder where presynaptic calcium channels are subjected to autoimmune destruction which causes fewer neurotransmitter vesicles to be exocytosed. This causes smaller EPPs due to less vesicles being released. Often the smaller EPPs do not reach threshold which causes muscle weakness and fatigue in patients. Many animals use neurotoxins to defend themselves and kill prey. Tetrodotoxin is a poison found in the certain poisonous fishes such as pufferfish and triggerfish which blocks the sodium ion channels and prevents an action potential on the postsynaptic membrane. Tetraethylammonium found in insects blocks potassium channels. Alpha neurotoxin found in snakes binds to acetylcholine receptors and prevents acetylcholine from binding. Alpha-latrotoxin found in black widow spiders causes a massive influx of calcium at the axon terminal and leads to an overflow of neurotransmitter release. Botulinum toxin produced by the bacteria Clostridium botulinum is the most powerful toxic protein. It prevents release of acetylcholine at the neuromuscular junction by inhibiting docking of the neurotransmitter vesicles.
See also
[edit]References
[edit]- ^ Boron, W.; Boulpaep, E. (2012). Medical Physiology. Philadelphia, PA: Saunders, Elsevier inc. p. 224. ISBN 978-0-8089-2449-4.
- ^ Kimura Y; Oda Y; Deguchi T; Higashida H (1992). "Enhanced acetylcholine secretion in neuroblastoma X glioma hybrid NG108-15 cells transfected with rat choline-acetyltransferase CDNA". FEBS Letters. 314 (3): 409–412. Bibcode:1992FEBSL.314..409K. doi:10.1016/0014-5793(92)81516-O. PMID 1468577. S2CID 4956377.
- ^ Lin S, Landmann L, Ruegg MA, Brenner HR (2008). "The role of nerve-versus muscle-derived factors in mammalian neuromuscular junction formation" (PDF). Journal of Neuroscience. 28 (13): 3333–3340. doi:10.1523/JNEUROSCI.5590-07.2008. PMC 6670584. PMID 18367600. S2CID 18659773.
- ^ Cole RN, Reddel SW, Gervasio OL, Phillips WD (2008). "Anti-MuSK patient antibodies disrupt the mouse neuromuscular junction". Annals of Neurology. 63 (6): 782–789. doi:10.1002/ana.21371. PMID 18384168. S2CID 205340971.
- ^ Teressa G, Prives J (2008). "Cell culture-based analysis of postsynaptic membrane assembly in muscle cells". Biological Procedures Online. 10 (1): 58–65. doi:10.1251/bpo143. PMC 2683546. PMID 19461953.
- ^ Van Lunteren E, Moyer M (2005). "Modulation of biphasic reate of end-plate potential recovery in rat diaphragm". Muscle & Nerve. 31 (3): 321–330. doi:10.1002/mus.20245. PMID 15654692. S2CID 31071429.
- ^ a b Takeda T, Sakata A, Matsuoka T (1999). "Fractal dimensions in the occurrence of miniature end-plate potential in a vertebrate neuromuscular junction". Progress in Neuro-Psychopharmacology & Biological Psychiatry. 23 (6): 1157–1169. doi:10.1016/S0278-5846(99)00050-0. PMID 10621955. S2CID 30988488.
- ^ Sellin LC, Molgo J, Thornquist K, Hansson B, Thesleff S (1996). "On the possible origin of giant or slow-rising miniature end plate potentials at the neuromuscular junction". Pflügers Archiv: European Journal of Physiology. 431 (3): 325–334. doi:10.1007/BF02207269. PMID 8584425. S2CID 8748384.
- ^ Partanen J (1999). "End plate spikes in the human electromyogram. Revision of the fusimotor theory". Journal of Physiology-Paris. 93 (1–2): 155–166. doi:10.1016/S0928-4257(99)80146-6. PMID 10084719. S2CID 4961877.
- ^ Purves D, Augustine G, et al. "Electrical Signals of Nerve Cells." Neuroscience. Sinauer Associates, Inc: Sunderland, Massachusetts, 2008. 25-39.
- ^ Prives J, Professor of Pharmacology, State University of New York at Stony Brook. Interviewed by Pierre Watson. 2008-11-18.
- ^ Kohara N, Lin TS, Fukudome T, Kimura J, Sakamoto T, et al. (2002). "Pathophysiology of weakness in a patient with congenital end-plate acetylcholinesterase deficiency". Muscle & Nerve. 25 (4): 585–592. doi:10.1002/mus.10073. PMID 11932977. S2CID 45891411.
- ^ Zalewska E, Hausmanowa-Petrusewicz I, Stahlberg E (2004). "Modeling studies on irregular motor unit potentials". Clinical Neurophysiology. 115 (3): 543–556. doi:10.1016/j.clinph.2003.10.031. PMID 15036049. S2CID 43828995.