
Community hub
Recent from talks
Contribute something
Nothing was collected or created yet.
Henry reaction
View on WikipediaThis article may be too technical for most readers to understand. (May 2019) |
| Henry reaction | |
|---|---|
| Named after | Louis Henry |
| Reaction type | Coupling reaction |
| Identifiers | |
| Organic Chemistry Portal | henry-reaction |
| RSC ontology ID | RXNO:0000086 |
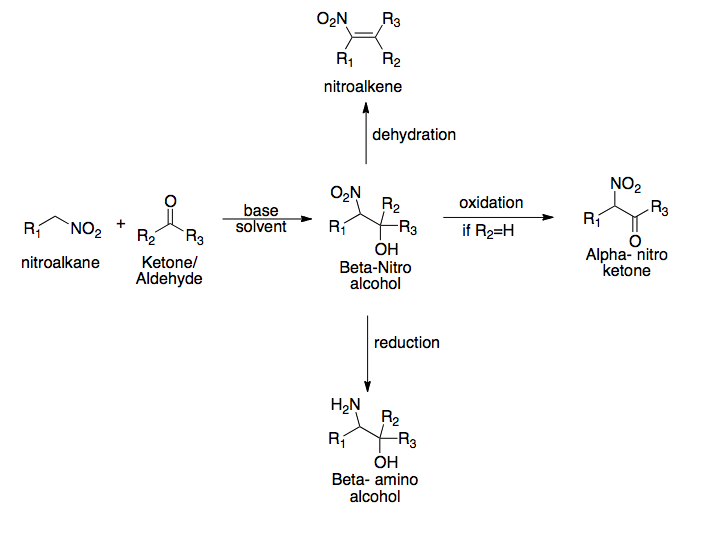
The Henry reaction is a classic carbon–carbon bond formation reaction in organic chemistry. Discovered in 1895 by the Belgian chemist Louis Henry (1834–1913), it is the combination of a nitroalkane and an aldehyde or ketone in the presence of a base to form β-nitro alcohols.[1][2][3] This type of reaction is also referred to as a nitroaldol reaction (nitroalkane, aldehyde, and alcohol). It is nearly analogous to the aldol reaction that had been discovered 23 years prior that couples two carbonyl compounds to form β-hydroxy carbonyl compounds known as "aldols" (aldehyde and alcohol).[2][4] The Henry reaction is a useful technique in the area of organic chemistry due to the synthetic utility of its corresponding products, as they can be easily converted to other useful synthetic intermediates. These conversions include subsequent dehydration to yield nitroalkenes, oxidation of the secondary alcohol to yield α-nitro ketones, or reduction of the nitro group to yield β-amino alcohols.

Many of these uses have been exemplified in the syntheses of various pharmaceuticals including the β-blocker (S)-propranolol,[5][6] the HIV protease inhibitor Amprenavir (Vertex 478), and construction of the carbohydrate subunit of the anthracycline class of antibiotics, L-Acosamine.[6] The synthetic scheme of the L-Acosamine synthesis can be found in the Examples section of this article.
Mechanism
[edit]The Henry reaction begins with the deprotonation of the nitroalkane on the α-carbon position forming a nitronate. The pKa of most nitroalkanes in DMSO is approximately 17.[7][8] Although this structure is nucleophilic both at the deprotonated carbon and at the oxy-anions of the nitro group,[9] the observed result is of the carbon attacking the carbonyl compound. The resulting β-nitro alkoxide is protonated by the conjugate acid of the base that originally deprotonated the nitroalkyl structure, giving the respective β-nitro alcohol as product.

All steps of the Henry reaction are reversible. This is due to the lack of a committed step in the reaction to form product. It is for this reason that research has been geared towards modifications to drive the reaction to completion.[2][3] More information about this can be found in the modification section of this article.
Stereochemical course
[edit]The figure below illustrates one of the commonly accepted models for stereoselection without any modification to the Henry reaction. In this model, stereoselectivity is governed by the size of the R groups in the model (such as a carbon chain), as well as by a transition state that minimizes dipole by orienting the nitro group and carbonyl oxygen anti each other (on opposite sides of the molecule). The R groups play a role in the transition state of the Henry reaction: the larger the R groups on each of the substrates, the more they will tend to orient themselves away from each other (commonly referred to as steric effects). [3][10]

Due to the reversibility of the reaction and the tendency for easy epimerization of the nitro-substituted carbon atom (among a number of factors), the Henry reaction will typically produce a mixture of enantiomers or diastereomers. It is for this reason that explanations for stereoselectivity remain scarce without some modification of the reaction.[3] In recent years, research focus has shifted toward modifications of the Henry reaction to overcome this synthetic challenge.

The first example of an enantioselective nitroaldol reaction was reported in 1992 using Shibasaki catalysts.[11] One of the most frequently employed methods for inducing enantio- or diastereoselectivity in the Henry reaction is the use of chiral metal catalysts, in which the nitro group and carbonyl oxygen coordinate to a metal that is bound to a chiral organic molecule. Some metals that have been used include zinc, cobalt, copper, magnesium, and chromium.[12] A depiction of this coordination is illustrated above.
General Features
[edit]One of the many features of the Henry reaction that makes it synthetically attractive is that it utilizes only a catalytic amount of base to drive the reaction. Additionally a variety of bases can be used including ionic bases such as alkali metal hydroxides, alkoxides, carbonates, and sources of fluoride anion (e.g. TBAF) or nonionic organic amine bases including TMG, DBU, DBN, and PAP. The base and solvent used do not have a large influence on the overall outcome of the reaction.[2]
Limitations
[edit]One of the main drawbacks of the Henry reaction is the potential for side reactions throughout. Aside from the inherent reversibility of the reaction (or "retro–Henry") that can prevent the reaction from proceeding, the β-nitro alcohol also has the potential to undergo dehydration. For sterically hindered substrates, it is also possible for a base-catalyzed self-condensation (Cannizzaro reaction) to occur. A general scheme of the Cannizzaro reaction is depicted below.[2]

Modifications
[edit]There have been a series of modifications made to the Henry reaction. Of these some of the most important include employing high-pressure and sometimes solvent free conditions to improve chemo- and regioselectivity[2] and chiral metal catalysts to induce enantio-or diastereoselectivity.[12] The aza-Henry reaction is also used to produce nitroamines and can be a reliable synthetic route for the synthesis of vicinal diamines.[13]
Perhaps one of the most synthetically useful modifications to the Henry reaction is the use of an organocatalyst.[2][12][14] The catalytic cycle is shown below.

Benjamin List described that while this is a broad explanation, his brief review illustrates that this is a plausible mechanistic explanation for almost all reactions that involve an organocatalyst. An example of this type of reaction is illustrated in the Examples section of this article.
In addition to the previously mentioned modifications to the Henry reaction there are a variety of others. This includes the conversion of unreactive alkyl nitro compounds to their corresponding dianions which will react faster with carbonyl substrates, reactions can be accelerated using PAP as base, utilization of the reactivity of aldehydes with α,α-doubly deprotonated nitroalkanes to give nitronate alkoxides that yield mainly syn-nitro alcohols once protonated, and finally generation of nitronate anions in which one oxygenatom on the nitro group is silyl-protected to yield anti-β-nitro alcohols in the presence of a fluoride anion source when reacted with an aldehyde.[2][3]
Examples
[edit]- Industrial Application
- In 1999, Menzel and coworkers developed a synthetic route to obtaining L-acosamine, the carbohydrate subunit of the anthracycline class of antibiotics:[6][15]

Acosamine scheme

- Industrial Application
- An enantioselective aldol addition product can be obtained in asymmetric synthesis by reaction of benzaldehyde with nitromethane and the a catalyst system consisting of zinc triflate as a Lewis acid, diisopropylethylamine (DIPEA), and N-methylephedrine (NME) as and as a chiral ligand.[16] A diastereoselective variation of this reaction is depicted below.[17]
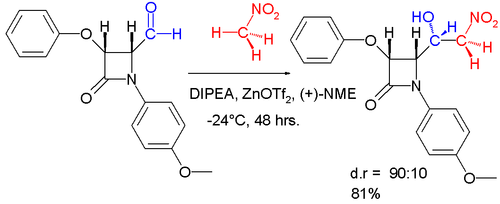
Henry reaction application

- Total Synthesis
- In 2005, Barua and coworkers completed the total synthesis of the potent aminopeptidase inhibitor, (–)-bestatin, in an overall yield of 26% overall yield employing Shibasaki's asymmetric Henry reaction as the key step. (illustrated below)[6][18]
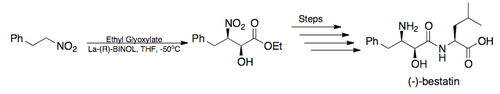
Bestatin

- Organocatalysis
- In 2006, Hiemstra and coworkers explored the use of quinine derivatives as asymmetric catalysts for the reaction between aromatic aldehydes and nitromethane. Through the use of particular derivatives, they were able to induce direct enantioselection through the use of the proper catalyst.[19]

Chinchura

- Biocatalysis
- In 2006, Purkarthofer et al. found that (S)-hydroxynitrile lyase from Hevea brasiliensis catalyzes the formation of (S)-β-nitro alcohols.[20] In 2011, Fuhshuku and Asano showed that the (R)-selective hydroxynitrile lyase from Arabidopsis thaliana could catalyze the synthesis of (R)-β-nitro alcohols from nitromethane and aromatic aldehydes.[21]
References
[edit]- ^ Henry, Louis (1895). "Formation synthétique d'alcools nitrés" [Synthetic formation of nitrated alcohols]. Comptes rendus. 120: 1265–1268.
- ^ a b c d e f g h Kurti, L.; Czako, B. (2005). Strategic Applications of Named Reactions in Organic Synthesis. Burlington, MA: Elsevier Academic Press. pp. 202–203. ISBN 978-0-12-369483-6.
- ^ a b c d e Noboro, Ono (2001). The Nitro Group in Organic Synthesis. New York, NY: Wiley-VCH. pp. 30–69. ISBN 978-0-471-31611-4.
- ^ Wurtz, M.A. (1872). "Sur un aldéhyde-alcool". Bull. Soc. Chim. Fr. 17: 436–442.
- ^ Sasai, H., Suzuki, T., Itoh, N., Arai, S., Shibasaki, M. (1993). "Catalytic Asymmetric Nitroaldol Reaction: an efficient synthesis of (s) propranolol using the lanthenum binaphthol complex". Tetrahedron Letters. 34 (52): 855–858. doi:10.1016/0040-4039(93)89031-K.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ^ a b c d Luzzio, F.A. (2001). "The Henry Reaction: recent examples". Tetrahedron. 57 (22): 915–945. doi:10.1002/chin.200122233.
- ^ Reich, Hans. "Bordwell pKa table: "Nitroalkanes"". University of Wisconsin Chemistry Department. Retrieved 17 January 2016.
- ^ Matthews, Walter; et al. (1975). "Equilibrium acidities of carbon acids. VI. Establishment of an absolute scale of acidities in dimethyl sulfoxide solution". Journal of the American Chemical Society. 97 (24): 7006. Bibcode:1975JAChS..97.7006M. doi:10.1021/ja00857a010.
- ^ Bersohn, Malcolm (1961). "C versus O Alkylation in the Case of a Stable Cation". J. Am. Chem. Soc. 83 (9): 2136–2138. Bibcode:1961JAChS..83.2136B. doi:10.1021/ja01470a022.
- ^ Begona, L., Arrieta, A., Morao, I., Cossio, F.P. (1997). "Ab Initio Models for the Nitroaldol (Henry) Reaction". Chem. Eur. J. 3 (1): 20–28. doi:10.1002/chem.19970030105.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ^ Sasai, Hiroaki; Suzuki, Takeyuki; Arai, Shigeru; Arai, Takayoshi; Shibasaki, Masakatsu (1 May 1992). "Basic character of rare earth metal alkoxides. Utilization in catalytic carbon-carbon bond-forming reactions and catalytic asymmetric nitroaldol reactions". Journal of the American Chemical Society. 114 (11): 4418–4420. Bibcode:1992JAChS.114.4418S. doi:10.1021/ja00037a068.
- ^ a b c List et al. described this process as the organocatalyst functioning as Lewis acid or base or Brønsted acid or base.
- ^ Westermann, B. (2003). "Asymmetric catalytic aza-Henry reactions leading to 1,2-diamines and 1,2-diaminocarboxylic acids". Angew. Chem. Int. Ed. Engl. 42 (2): 151–153. doi:10.1002/anie.200390071. PMID 12532343.
- ^ Seayad, J., List, B. (2005). "Asymmetric organocatalysis". Org. Biomol. Chem. 3 (5): 719–724. doi:10.1039/b415217b. PMID 15731852.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ^ Menzel, A., Ohrlein, R., Griesser, H., Wehner, V., Jager, V. (1999). "A Short Synthesis of L-Acosamine Based on Nitroaldol Addition (Henry Reaction). Analysis of the Key Step Concerning Solvent and Temperature Effects". Synthesis. 9 (45): 1691–1702. doi:10.1002/chin.199945325.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ^ Palomo, Claudio; Oiarbide, Mikel; Laso, Antonio (2005). "Enantioselective Henry Reactions under Dual Lewis Acid/Amine Catalysis Using Chiral Amino Alcohol Ligands". Angewandte Chemie. 44 (25): 3881–3884. doi:10.1002/anie.200463075. PMID 15892142.
- ^ Alcaide, Benito; Almendros, Pedro; Luna, Amparo; Paz de Arriba, M.; Rosario Torresc, M. (2007). "Organocatalyzed diastereoselective Henry reaction of enantiopure 4-oxoazetidine-2-carbaldehydes" (PDF). Arkivoc. 2007 (iv): 285–296. doi:10.3998/ark.5550190.0008.425.
- ^ Gogoi, N., Boruwa, J., Barua, N.C. (2005). "A total synthesis of (–)-bestatin using Shibasaki's asymmetric Henry reaction". Tetrahedron Letters. 46 (44): 7581–7582. doi:10.1016/j.tetlet.2005.08.153.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ^ Marcelli, T., van der Haas, R., van Maarseveen, J.H., Hiemstra, H. (2006). "Asymmetric Organocatalytic Henry Reaction". Angew. Chem. Int. Ed. 45 (6): 929–931. doi:10.1002/anie.200503724. PMID 16429453.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ^ Purkarthofer, T., Gruber, K., Gruber-Khadjawi, M., Waich, K., Skranc, W., Mink, D. and Griengl, H. (2006). "A Biocatalytic Henry Reaction—The Hydroxynitrile Lyase from Hevea brasiliensis Also Catalyzes Nitroaldol Reactions". Angewandte Chemie. 45 (21): 3454–3456. doi:10.1002/anie.200504230. PMID 16634109.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ^ Fuhshuku K, Asano Y (2011). "Synthesis of (R)-β-nitro alcohols catalyzed by R-selective hydroxynitrile lyase from Arabidopsis thaliana in the aqueous-organic biphasic system". J. Biotechnol. 153 (3–4): 153–159. doi:10.1016/j.jbiotec.2011.03.011. PMID 21439333.
External links
[edit] Media related to Henry reaction at Wikimedia Commons
Media related to Henry reaction at Wikimedia Commons
Henry reaction
View on GrokipediaBackground
Definition and Overview
The Henry reaction, also known as the nitroaldol reaction, is a classic organic transformation that involves the base-catalyzed addition of a nitroalkane to an aldehyde or ketone, resulting in the formation of β-nitro alcohols.[5][6] This carbon-carbon bond-forming process is analogous to the aldol reaction but utilizes the nucleophilic nitronate anion derived from the nitroalkane instead of an enolate.[5] Discovered in 1895 by Belgian chemist Louis Henry, the reaction exemplifies how the acidity of nitro compounds enables efficient nucleophilic additions to carbonyls.[7] The general equation for the Henry reaction is represented as: where R and R' are alkyl or aryl substituents, and the base facilitates deprotonation of the nitroalkane at the α-position.[5][6] Nitroalkanes exhibit enhanced acidity at this position (pKa values typically ranging from 8 to 12) due to resonance stabilization of the resulting nitronate anion by the electron-withdrawing nitro group, making them suitable nucleophiles under mild basic conditions.[8] The products of the Henry reaction contain a characteristic β-hydroxy nitro functionality, which imparts versatility for subsequent synthetic manipulations, such as dehydration to nitroalkenes or conversion to carbonyl compounds via the Nef reaction.[5][9] This structural motif has made the reaction a valuable tool in organic synthesis for constructing complex molecules, particularly in the synthesis of pharmaceuticals and natural products.[6]Historical Development
The Henry reaction was discovered in 1895 by Belgian chemist Louis Henry (1834–1913), who first described the base-catalyzed condensation of nitromethane with aldehydes to afford β-nitro alcohols. In his initial experiments, Henry employed potassium carbonate as the base to facilitate the addition of nitromethane to acetaldehyde, yielding 1-nitro-2-propanol among other products, thereby establishing a new method for carbon-carbon bond formation analogous to cyanohydrin synthesis. This discovery stemmed from Henry's broader investigations into nitroparaffins and their reactivity, highlighting the acidic nature of the α-hydrogen in nitromethane.[10] Henry's early publications detailed these findings, beginning with the foundational 1895 communication in Comptes rendus hebdomadaires des séances de l'Académie des sciences titled "Formation synthétique d'alcools nitrés," which outlined the preparation and properties of the nitro alcohol products. Follow-up works through the late 1890s and into the 1900s, published in the same journal and Bulletin de la Société chimique de France, explored variations with other nitroalkanes and aldehydes, confirming the general applicability of the process and characterizing derivatives such as acetates and halides of the β-nitro alcohols. These efforts solidified the reaction's role in organic synthesis during the early 20th century.[10] Named the "Henry reaction" shortly after its inception to honor the discoverer, the process retained this designation in much of the classical literature. However, by the post-1950s era, it gained prominence as the "nitroaldol reaction" in recognition of its mechanistic parallel to the aldol addition, a shift evident in comprehensive organic chemistry texts and reviews that emphasized its nucleophilic addition character.[11] Significant milestones advanced the reaction's scope thereafter. Although Henry attempted extensions to ketones in 1895 with modest results, practical applications with ketones emerged in the 1920s through refined base catalysis and conditions, enabling broader substrate compatibility. The 1990s introduced asymmetric catalysis, with Shibasaki and coworkers reporting the first enantioselective variant in 1992 using a chiral heterobimetallic lithium-lanthanum-BINOL complex, achieving up to 98% enantiomeric excess in additions to aromatic aldehydes and paving the way for stereoselective syntheses.Reaction Mechanism
Formation of Nitronate Intermediate
The formation of the nitronate intermediate constitutes the initial step in the Henry reaction, wherein a base abstracts the α-proton from a nitroalkane to generate a resonance-stabilized anion that serves as the nucleophile.[12] The nitro group enhances the acidity of the α-hydrogen through electron withdrawal, resulting in pKa values of approximately 10 for simple nitroalkanes like nitromethane, facilitating deprotonation by a variety of bases.[13] Typical bases employed include alkoxides such as sodium ethoxide and amines like triethylamine or 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), which act as proton acceptors to yield the nitronate anion. For anionic bases (e.g., alkoxides): . For neutral bases (e.g., amines): , where represents an alkyl substituent and (or ) denotes the base.[5] This process is reversible, establishing an equilibrium that is governed by the base's strength relative to the nitroalkane's acidity and modulated by solvent polarity, with protic solvents like ethanol favoring partial deprotonation while aprotic ones like dimethyl sulfoxide promote more complete anion formation.[14] The existence and structure of the nitronate anion in solution have been substantiated through spectroscopic techniques, notably H and C NMR studies, which reveal characteristic chemical shifts for the α-carbon and surrounding protons indicative of the delocalized negative charge on the nitro group oxygens.[15] These observations confirm the anion's aci-nitro form () as the predominant tautomer under basic conditions.[15]Nucleophilic Addition and Protonation
In the Henry reaction, the nucleophilic addition step involves the attack of the nitronate anion on the electrophilic carbonyl carbon of an aldehyde, resulting in the formation of a new carbon-carbon bond and an alkoxide intermediate. This process is represented by the equation: The nitronate acts as a carbanion equivalent, stabilized by the adjacent nitro group, enabling efficient addition to the polarized carbonyl.[16] The transition state for this addition typically adopts zigzag or extended conformations, allowing the nitronate carbon to approach the carbonyl in a manner that minimizes electronic repulsion while forming the partial bonds. Steric factors significantly influence this step, as increased bulk around the aldehyde's α-position or the nitronate's substituents can raise the activation barrier, reducing the reaction rate.[17] The nucleophilic addition is generally the rate-determining step, governed by the kinetics of the bimolecular collision between the nitronate and aldehyde, with steric hindrance often dictating overall efficiency. Subsequent protonation of the alkoxide intermediate occurs during aqueous workup, where the negatively charged oxygen accepts a proton to afford the neutral β-nitroalcohol product. This protonation is rapid and typically requires no additional catalysts under standard conditions.[16][6]Stereochemistry
Diastereoselectivity
The diastereoselectivity of the Henry reaction governs the relative configuration of the β-nitroalcohol products, yielding syn and anti diastereomers arising from the nucleophilic addition of the nitronate to the aldehyde carbonyl. In non-chelated conditions, such as base-catalyzed reactions without metal coordination, the process typically proceeds through an open transition state, resulting in low to moderate diastereoselectivity with a preference for the anti isomer; for example, control experiments using nitroethane and benzaldehyde yield syn:anti ratios of 1:1.4.[18] Metal-coordinated variants, particularly those involving Lewis acids like Mg or Zn salts, enable diastereocontrol via chelation of both the nitronate oxygen and the aldehyde oxygen, forming a Zimmerman-Traxler-like six-membered chair transition state that favors the syn product. This chelated model minimizes steric repulsion between substituents, promoting the syn geometry, as observed in various Lewis acid systems. Classic studies demonstrate variable diastereomeric ratios (dr) depending on reaction conditions and substrates, ranging from nearly 1:1 in uncatalyzed or weakly coordinated systems to high syn:anti with effective Lewis acid chelation; for instance, Ca²⁺-crosslinked alginate catalysts with nitroethane show minor dr (syn:anti ≈ 1.3:1) but a trend toward syn enhancement compared to non-metal controls.[18] Substrate sterics significantly influence selectivity, with bulky R groups on the aldehyde or nitroalkane favoring anti products by destabilizing the chelated syn transition state due to 1,3-diaxial interactions, whereas less hindered substrates align better with syn-favoring chelation.[20]Enantioselectivity in Asymmetric Variants
The development of enantioselective variants of the Henry reaction has significantly expanded its utility in organic synthesis by enabling the preparation of chiral β-nitroalcohols with high enantiomeric excess (ee). Pioneering work in the 1990s by Shibasaki and coworkers introduced binaphthol (BINOL)-based rare earth metal catalysts, marking the first catalytic asymmetric Henry reactions. In their seminal 1992 report, a lanthanum-BINOL complex catalyzed the addition of nitromethane to aldehydes such as benzaldehyde, affording the product in 93% yield and 92% ee under mild conditions.[21] Subsequent refinements using heterobimetallic BINOL complexes, such as La-Li-BINOL systems, improved enantioselectivity to >95% ee for a range of aromatic and aliphatic aldehydes, with yields often exceeding 90%.[11] These catalysts operate through cooperative Lewis acid activation of the carbonyl and Brønsted base deprotonation of the nitroalkane, establishing a benchmark for metal-mediated asymmetric control.[22] Organocatalytic methods emerged in the early 2000s, providing metal-free alternatives for enantioselective Henry reactions. Early examples included chiral phase-transfer catalysts derived from cinchona alkaloids, as reported by Ooi, Maruoka, and coworkers in 2003, which delivered up to 90% ee in the addition of nitroalkanes to aromatic aldehydes with good yields (80-95%). Proline-derived organocatalysts, particularly sulfonamide derivatives, were later adapted for Henry reactions, achieving enantioselectivities of 82-96% ee in benchmark additions of nitromethane to aliphatic aldehydes, often in 70-90% yields.[23] These approaches prioritize simple, scalable conditions and broad substrate tolerance, complementing metal-based systems. Recent advances as of 2025 include Cu-diamine ligand systems that achieve >95% ee in asymmetric Henry reactions for synthesizing pharmaceutical intermediates like linezolid precursors, expanding scope to challenging substrates.[24] Enantioselectivity in these asymmetric variants is primarily governed by bifunctional catalysis mechanisms, where the catalyst simultaneously activates both substrates through hydrogen bonding and coordination. In BINOL-metal systems, the chiral environment enforces si or re face selectivity via steric differentiation around the metal center.[22] For organocatalytic variants, bifunctional H-bond donors like thioureas or sulfonamides engage the nitro group as an H-bond acceptor and the carbonyl oxygen, orienting the nitronate for approach from one enantiotopic face; computational models confirm this dual activation enhances ee by stabilizing the transition state leading to the (R) or (S) product depending on catalyst configuration.[23] In the benchmark reaction of nitromethane with benzaldehyde, such models predict and achieve >90% ee, underscoring the role of precise H-bonding in face selection.[25]Scope and Selectivity
Substrate Compatibility
The Henry reaction exhibits broad compatibility with various aldehydes as electrophilic partners, with aromatic aldehydes such as benzaldehyde being particularly effective due to their moderate reactivity, leading to high yields of β-nitroalcohols under standard conditions.[26] Aliphatic aldehydes, including straight-chain examples like pentanal and octanal, are also viable substrates, though they often require optimized conditions to achieve comparable efficiency owing to higher reactivity and potential side pathways.[27] Ketones participate less frequently in the reaction because of their lower electrophilicity, but activated variants such as α-ketoesters react successfully to afford the corresponding nitroaldol products in good yields.[26] Primary nitroalkanes, exemplified by nitromethane, serve as the most commonly employed nucleophilic components, providing unsubstituted β-nitroalcohols with excellent efficiency across a range of carbonyl partners.[5] Nitroethane, another primary nitroalkane, enables the synthesis of 2-methyl-substituted β-nitroalcohols and is widely utilized for introducing alkyl groups at the α-position.[28] Secondary nitroalkanes, such as 2-nitropropane, can participate but typically deliver lower yields and necessitate longer reaction times compared to their primary counterparts due to steric hindrance in forming the nitronate anion. The reaction demonstrates good tolerance for a variety of functional groups on the aldehyde substrates, including ethers (e.g., methoxy), halides (e.g., chloro and fluoro), and electron-withdrawing groups like cyano and nitro, allowing for the preparation of functionalized β-nitroalcohols without interference.[27] However, substrates bearing acidic protons or those sensitive to strong basic conditions, such as carboxylic acids, are generally incompatible as they may undergo deprotonation or decomposition under the reaction's basic environment.[26] In terms of expanded scope, α,β-unsaturated aldehydes can undergo the Henry reaction, primarily via 1,2-addition, though rare variants promote conjugate (1,4-) addition of nitroalkanes to yield γ-nitro carbonyl compounds.[28]Reaction Conditions and Catalysts
The classical Henry reaction employs mild basic conditions, typically utilizing 10-20% aqueous NaOH or sodium ethoxide (EtONa) as the base in polar solvents such as ethanol (EtOH) or tetrahydrofuran (THF) at temperatures ranging from 0 to 25 °C, with reaction times of 1 to 24 hours to afford β-nitro alcohols in good yields.[29] These conditions are suitable for simple substrates like aromatic aldehydes and nitromethane, often proceeding with excess nitroalkane to shift the equilibrium toward the product.[30] A variety of catalysts enhance the efficiency and selectivity of the Henry reaction beyond classical bases. Homogeneous amine bases, such as 1,4-diazabicyclo[2.2.2]octane (DABCO), promote rapid reactions under solvent-free or mild conditions, delivering high yields (up to 99%) in short times at room temperature. Heterogeneous catalysts, including anion-exchange resins like Amberlyst A-21, offer recyclability and facilitate ambient-temperature reactions in aprotic solvents, with yields exceeding 80% for aliphatic and aromatic aldehydes.[29] Metal-based catalysts, particularly copper (Cu) and nickel (Ni) salts coordinated with chiral ligands, enable chelation-controlled additions, achieving enantioselectivities up to 99.5% ee at 0 °C in THF.[31] Solvent choice significantly influences solubility and reaction kinetics in the Henry reaction. Polar protic solvents like methanol (MeOH) improve the solubility of nitroalkanes and bases, supporting efficient reactions for poorly soluble substrates with yields around 90%.[29] In contrast, polar aprotic solvents such as dimethyl sulfoxide (DMSO) accelerate rates by stabilizing the nitronate anion, often resulting in faster conversions (up to 50 times quicker than in protic media) and higher yields for electron-deficient aldehydes. For scale-up, the Henry reaction maintains robust performance, yielding 70-95% for simple substrates under optimized classical or catalyzed conditions, as demonstrated in multigram syntheses using aqueous NaOH or Cu-based systems without significant loss in efficiency.[29]Limitations and Side Reactions
Common Challenges
One common challenge in the Henry reaction is the occurrence of side reactions, particularly the aldol self-condensation of aldehydes under basic conditions, which competes with the desired nitroaldol addition and reduces overall efficiency.[32] Additionally, β-nitro alcohols formed as products can undergo dehydration to nitroalkenes, particularly under acidic or heating conditions, or retro-Henry reversal under basic conditions, leading to unwanted byproducts and complicating reaction outcomes.[33] Yield limitations frequently arise with sterically hindered substrates, such as ortho-substituted benzaldehydes, where steric congestion impedes nucleophilic approach and results in low conversions, often below 50%. For instance, reactions involving ortho-alkoxybenzaldehydes without optimized conditions typically afford reduced yields. Purification of β-nitro alcohols poses significant difficulties due to their proneness to dehydration, which generates nitroalkenes during isolation or storage.[34] These compounds also exhibit instability in acidic media, where degradation can occur, further hindering handling and recovery.[35] Historically, prior to the 1980s, the Henry reaction suffered from poor stereocontrol, with classical methods providing little to no enantioselectivity, limiting its utility in asymmetric synthesis.[11]Strategies to Overcome Limitations
To suppress side reactions such as the dehydration to form nitroalkenes or retro-Henry reversal, employing an excess of the nitroalkane reactant shifts the equilibrium toward the desired β-nitroalcohol product, as the Henry reaction is reversible under basic conditions.[6] Additionally, conducting the reaction at low temperatures favors the nucleophilic addition pathway over elimination, which is thermodynamically driven at higher temperatures.[36] Yields in the Henry reaction can be improved through phase-transfer catalysis in biphasic systems, where tetrabutylammonium bromide (TBAB) facilitates the transfer of hydroxide ions from aqueous NaOH to the organic phase, enabling efficient deprotonation of nitroalkanes and achieving conversions up to 95% for various aldehydes.[37] Microwave irradiation further accelerates the process in solvent-free conditions, reducing reaction times from hours to minutes while enhancing yields to 80-99% by promoting uniform heating and minimizing side products.[38] For enhanced stereocontrol, particularly in diastereoselective variants, the addition of water as an additive modulates hydrogen bonding interactions in the transition state, leading to improved syn/anti ratios in reactions catalyzed by chiral copper complexes.[39] Recent advances post-2015 include computational mechanistic studies that elucidate the retro-Henry elimination pathway, such as density functional theory analyses of catalyst-substrate interactions in copper systems, aiding the rational design of catalysts to suppress reversibility and improve selectivity in asymmetric transformations.[40]Variations
Classical vs. Organocatalytic Approaches
The classical Henry reaction, first reported by Louis Henry in 1895, employs strong inorganic bases such as sodium hydroxide or milder organic bases like triethylamine to deprotonate nitroalkanes, enabling their addition to aldehydes and yielding β-nitro alcohols with broad substrate scope across aromatic and aliphatic carbonyls.[4] These conditions often achieve good yields of 70–95% for simple substrates like benzaldehyde and nitromethane, but the process typically produces racemic products with poor diastereoselectivity, limiting its utility in asymmetric synthesis, and may require elevated temperatures or protic solvents that can lead to side reactions like self-condensation.[5] From the 1890s through the late 1990s, this approach dominated due to its simplicity and effectiveness for non-stereoselective applications, though the use of stoichiometric bases generated significant inorganic waste.[41] In contrast, organocatalytic variants, emerging prominently in the 2000s, utilize metal-free chiral catalysts such as cinchona alkaloid derivatives (e.g., quinine thioureas) or bifunctional thioureas to activate both the nitroalkane and carbonyl through hydrogen bonding, enabling highly enantioselective additions under mild conditions.[1] Seminal work with C6'-modified cinchona alkaloids demonstrated enantioselectivities up to 99% ee and yields exceeding 90% for challenging ketone substrates at room temperature in non-coordinating solvents like toluene, while thiourea-based systems, such as guanidine-thiourea hybrids, provided syn-selective products with >95% ee for aldehyde-nitroalkane couplings.[42] These methods excel in diastereoselectivity (often >20:1 syn:anti) and compatibility with sensitive functional groups, addressing classical limitations without metal toxicity.[43] The shift toward organocatalytic approaches post-2000 reflects demands for green chemistry, as these catalysts operate under ambient conditions, reduce waste through low catalyst loadings (1–10 mol%), and allow catalyst recovery, minimizing environmental impact compared to classical methods that rely on non-recyclable bases.[44] Representative comparisons highlight these differences:| Aspect | Classical Approaches | Organocatalytic Approaches |
|---|---|---|
| Typical Yields | 70–95% (e.g., NaOH in EtOH/H₂O) | 80–99% (e.g., cinchona thiourea in PhMe) |
| Enantioselectivity | Racemic (0% ee) | Up to 99% ee |
| Conditions | Often 0–60°C, protic solvents, stoichiometric base | Room temperature, aprotic solvents, catalytic (1–10 mol%) |
| Environmental Impact | High waste from inorganic salts, potential toxicity | Metal-free, recyclable, lower E-factor |
Recent Modifications
Recent advancements in the Henry reaction since 2020 have focused on sustainable and efficient methodologies, including photocatalytic variants that leverage visible light to enable radical pathways. In 2021, cyanine-based organic dyes were introduced as metal-free photoredox catalysts for the aza-Henry reaction, a nitroalkane addition to imines, under near-infrared irradiation. These dyes, such as cy746, facilitate oxidative C-C bond formation between tetrahydroisoquinolines and nitromethane or other nitroalkanes, achieving yields up to 91% in DMSO solvent without harsh conditions. This approach highlights the potential of organic photocatalysts to activate nitro compounds via single-electron transfer, offering milder alternatives to traditional base-catalyzed processes.[49] Biocatalytic modifications have gained traction for their environmental benefits, particularly through enzyme engineering to enhance sustainability. A 2024 study demonstrated that the hydroxynitrile lyase from Bambusa multiplex (BmHNL) catalyzes the promiscuous asymmetric Henry reaction, adding nitromethane to various aldehydes to yield β-nitroalcohols with high enantioselectivity (>99% ee for (S)-enantiomers) and up to >99% conversion under aqueous conditions at ambient temperature. This enzyme also enables enantiocomplementary synthesis via retro-Henry reactions for (R)-enantiomers. These developments address limitations in substrate scope and stability, enabling greener synthesis of chiral building blocks for pharmaceuticals. Ongoing research emphasizes promiscuous enzyme activity for diastereoselective outcomes in biocatalytic C-C bond formation.[50] Flow chemistry adaptations have improved the scalability and safety of the Henry reaction, especially when handling explosive nitro compounds like nitromethane. These setups allow precise control of residence time and temperature, facilitating safe scale-up to multigram quantities without batch hazards. By integrating immobilized bases, the process supports recycling and reduces waste, making it suitable for industrial applications. Emerging metal-free multicomponent Henry-Michael cascades have expanded the reaction's utility for complex molecule assembly post-2023. A catalyst-free approach in 2023 enabled a sequential Michael addition followed by Henry reaction using functionalized vinylogous nucleophiles and nitroalkanes, constructing diastereomerically pure bicyclo[3.2.1]octanes in yields up to 92%.[51] This tandem process avoids metal catalysts, relying on inherent reactivity under mild heating, and has been applied to synthesize polycyclic frameworks. Further innovations include bifunctional metal-organic frameworks (MOFs) that promote four-step cascades incorporating Henry-Michael steps, though metal-free variants prioritize organocatalysts for sustainability in constructing functionalized nitro compounds.[52]Synthetic Applications
Key Examples in Total Synthesis
A prominent modern example is the total synthesis of oseltamivir (Tamiflu), an antiviral drug, featuring an asymmetric aza-Henry reaction (a variant of the Henry reaction involving imines) as a pivotal step for introducing the β-nitroamine functionality. Reported in 2010 by Ko et al., this azide-free route started from commercially available D-mannitol and proceeded in 18 steps, culminating in oseltamivir phosphate with an overall yield of 13.7%. The aza-Henry step, catalyzed by a chiral thiourea organocatalyst, delivered the key intermediate in 91% yield and 95% ee, which was then reduced with zinc in acetic acid to the corresponding β-amino alcohol, followed by deprotection and cyclization to the cyclohexene core. This synthesis demonstrated the scalability of asymmetric Henry variants for pharmaceutical targets, avoiding hazardous azides used in earlier routes.[53] In carbohydrate chemistry, the Henry reaction has facilitated the synthesis of nitrocyclitols during the 2000s and 2010s, particularly through intramolecular variants to form carbocyclic structures. For instance, a 2003 synthesis transformed D-glucose-derived nitroheptofuranoses into 1D-3-deoxy-3-hydroxymethyl-myo-inositols via an intramolecular Henry cyclization, proceeding in good yield with high diastereoselectivity due to the rigid furanose template.[54] The resulting nitro cyclitols were reduced using Raney nickel to afford the corresponding aminocyclitols, providing valuable scaffolds for glycosidase inhibitors. This strategy underscored the reaction's role in stereocontrolled ring construction and functional group interconversion in complex natural product analogs.Industrial and Pharmaceutical Uses
The Henry reaction plays a significant role in the pharmaceutical synthesis of β-blockers, where β-nitro alcohol intermediates derived from the reaction serve as key precursors for drugs such as (S)-metoprolol, (S)-toliprolol, and (S)-propranolol. These asymmetric variants, catalyzed by copper complexes with chiral ligands, achieve high enantioselectivity (>90% ee) and yields, enabling the production of optically pure therapeutic agents used in treating cardiovascular conditions.[55] Early applications in the 1980s and beyond highlighted the reaction's utility in constructing the nitro alcohol core, which is subsequently reduced and elaborated into the final β-adrenergic antagonists.[34] In industrial agrochemical production, the Henry reaction facilitates the synthesis of herbicide safeners like furilazole, a dichloroacetamide compound that protects crops such as maize from the phytotoxic effects of herbicides including isoxaflutole and sulfonylureas. The reaction between furfural and nitromethane, catalyzed by barium hydroxide, yields the intermediate α-(nitromethyl)-2-furanmethanol in 78.9% yield under optimized conditions, followed by nitro reduction and cyclization steps that support scalable manufacturing. This approach enhances herbicide efficacy by boosting crop detoxification enzymes like cytochrome P450 and glutathione S-transferases, contributing to higher agricultural yields while minimizing environmental impact. Cost-effectiveness is further improved through catalyst recycling, maintaining yields above 96% over multiple cycles.[56] A 2014 total synthesis of zanamivir, an anti-influenza neuraminidase inhibitor, incorporated the Henry reaction's anti-selective addition, catalyzed by CuBr₂ with a proline-derived ligand, producing β-nitro alcohols with 98% ee and 60% yield, integrated into a 13-step route from D-araboascorbic acid achieving 18% overall yield.[57] Reviews from 2021 emphasize the reaction's role in greener asymmetric syntheses for such scaffolds, supporting diversity-oriented approaches in antiviral discovery.[57] The Henry reaction's versatility enables efficient construction of nitro-functionalized motifs for large-scale diversity synthesis in both sectors, but challenges arise in purification at industrial scales due to byproduct formation and the need for extensive chromatography, as seen in aza-Henry scale-ups where yields drop without optimized flow processes. Continuous flow methodologies address these issues, allowing selective production of nitroaldols or derivatives over fixed-bed catalysts, enhancing throughput and cost-effectiveness for commercial viability.References
- https://onlinelibrary.wiley.com/doi/10.1002/1521-3757(20020301)114:5<889::AID-ANGE889>3.0.CO;2-8