
Community hub
Recent from talks
Contribute something
Nothing was collected or created yet.
Organocatalysis
View on Wikipedia
In organic chemistry, organocatalysis is a form of catalysis in which the rate of a chemical reaction is increased by an organic catalyst. This "organocatalyst" consists of carbon, hydrogen, sulfur and other nonmetal elements found in organic compounds.[3][4][5][6][7][8] Because of their similarity in composition and description, they are often mistaken as a misnomer for enzymes due to their comparable effects on reaction rates and forms of catalysis involved.
Organocatalysts which display secondary amine functionality can be described as performing either enamine catalysis (by forming catalytic quantities of an active enamine nucleophile) or iminium catalysis (by forming catalytic quantities of an activated iminium electrophile). This mechanism is typical for covalent organocatalysis. Covalent binding of substrate normally requires high catalyst loading (for proline-catalysis typically 20–30 mol%). Noncovalent interactions such as hydrogen-bonding facilitates low catalyst loadings (down to 0.001 mol%).
Organocatalysis offers several advantages. There is no need for metal-based catalysis thus making a contribution to green chemistry. In this context, simple organic acids have been used as catalyst for the modification of cellulose in water on multi-ton scale.[9] When the organocatalyst is chiral an avenue is opened to asymmetric catalysis; for example, the use of proline in aldol reactions is an example of chirality and green chemistry.[10] Organic chemists David MacMillan and Benjamin List were both awarded the 2021 Nobel Prize in chemistry for their work on asymmetric organocatalysis.[11]
Introduction
[edit]Regular achiral organocatalysts are based on nitrogen such as piperidine used in the Knoevenagel condensation.[12] DMAP used in esterifications[13] and DABCO used in the Baylis-Hillman reaction.[14] Thiazolium salts are employed in the Stetter reaction.[15] These catalysts and reactions have a long history but current interest in organocatalysis is focused on asymmetric catalysis with chiral catalysts, called asymmetric organocatalysis or enantioselective organocatalysis. A pioneering reaction developed in the 1970s is called the Hajos–Parrish–Eder–Sauer–Wiechert reaction. Between 1968 and 1997, there were only a few reports of the use of small organic molecules as catalysts for asymmetric reactions (the Hajos–Parrish reaction probably being the most famous), but these chemical studies were viewed more as unique chemical reactions than as integral parts of a larger, interconnected field.[16]

In this reaction, naturally occurring chiral proline is the chiral catalyst in an aldol reaction. The starting material is an achiral triketone and it requires just 3% of proline to obtain the reaction product, a ketol in 93% enantiomeric excess. This is the first example of an amino acid-catalyzed asymmetric aldol reaction.[17][18]
The asymmetric synthesis of the Wieland-Miescher ketone (1985) is also based on proline and another early application was one of the transformations in the total synthesis of Erythromycin by Robert B. Woodward (1981).[19] A mini-review digest article focuses on selected recent examples of total synthesis of natural and pharmaceutical products using organocatalytic reactions.[20]
Many chiral organocatalysts are an adaptation of chiral ligands (which together with a metal center also catalyze asymmetric reactions) and both concepts overlap to some degree.
A breakthrough in the field of organocatalysis came in 1997 when Yian Shi reported the first general, highly enantioselective organocatalytic reaction with the catalytic asymmetric epoxidation of trans- and trisubstituted olefins with chiral dioxiranes.[21] Since that time, several different types of reactions have been developed.
Organocatalyst classes
[edit]Organocatalysts for asymmetric synthesis can be grouped in several classes:
- Biomolecules: proline, phenylalanine. Secondary amines in general.[22] The cinchona alkaloids, certain oligopeptides.
- Synthetic catalysts derived from biomolecules.
- Hydrogen bonding catalysts, including TADDOLS, derivatives of BINOL such as NOBIN, and organocatalysts based on thioureas
- Triazolium salts as next-generation Stetter reaction catalysts
Examples of asymmetric reactions involving organocatalysts are:
Proline
[edit]Imidazolidinone organocatalysis
[edit]
Imidazolidinones are catalysts for many transformations such as asymmetric Diels-Alder reactions and Michael additions. Chiral catalysts induce asymmetric reactions, often with high enantioselectivities. This catalyst works by forming an iminium ion with carbonyl groups of α,β-unsaturated aldehydes (enals) and enones in a rapid chemical equilibrium. This iminium activation is similar to activation of carbonyl groups by a Lewis acid and both catalysts lower the substrate's LUMO:[25][26]

The transient iminium intermediate is chiral which is transferred to the reaction product via chiral induction. The catalysts have been used in Diels-Alder reactions, Michael additions, Friedel-Crafts alkylations, transfer hydrogenations and epoxidations.
One example is the asymmetric synthesis of the drug warfarin (in equilibrium with the hemiketal) in a Michael addition of 4-hydroxycoumarin and benzylideneacetone:[27]

A recent exploit is the vinyl alkylation of crotonaldehyde with an organotrifluoroborate salt:[28]

For other examples of its use: see organocatalytic transfer hydrogenation and asymmetric Diels-Alder reactions.
Thiourea organocatalysis
[edit]A large group of organocatalysts incorporate the urea or the thiourea moiety. These catalytically effective (thio)urea derivatives termed (thio)urea organocatalysts provide explicit double hydrogen-bonding interactions to coordinate and activate H-bond accepting substrates.[29]
Their current uses are restricted to asymmetric multicomponent reactions, including those involving Michael addition, asymmetric multicomponent reactions for the synthesis of spirocycles, asymmetric multicomponent reactions involving acyl Strecker reactions, asymmetric Petasis reactions, asymmetric Biginelli reactions, asymmetric Mannich reactions, asymmetric aza-Henry reactions, and asymmetric reductive coupling reactions.[30]
References
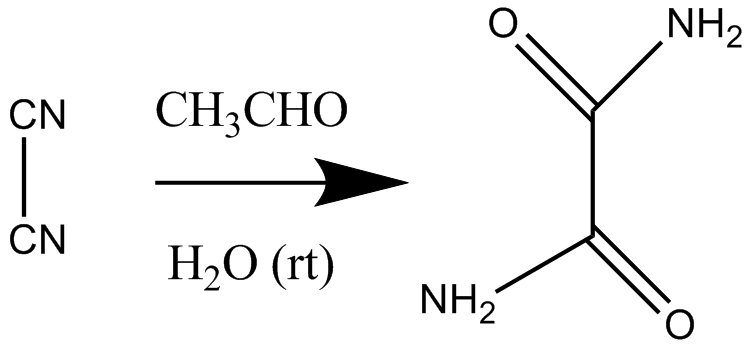
[edit]- ^ Justus von Liebig, Justus (1860). "Ueber die Bildung des Oxamids aus Cyan". Annalen der Chemie und Pharmacie. 113 (2): 246–247. doi:10.1002/jlac.18601130213.
- ^ W. Langenbeck (1929). "Über organische Katalysatoren. III. Die Bildung von Oxamid aus Dicyan bei Gegenwart von Aldehyden". Liebigs Ann. 469: 16–25. doi:10.1002/jlac.19294690103.
- ^ Berkessel, A.; Groeger, H. (2005). Asymmetric Organocatalysis. Weinheim: Wiley-VCH. ISBN 978-3-527-30517-9.
- ^ Special Issue: List, Benjamin (2007). "Organocatalysis". Chem. Rev. 107 (12): 5413–5883. doi:10.1021/cr078412e.
- ^ Peter I. Dalko; Lionel Moisan (2004). "In the Golden Age of Organocatalysis". Angew. Chem. Int. Ed. 43 (39): 5138–5175. doi:10.1002/anie.200400650. PMID 15455437.
- ^ Matthew J. Gaunt; Carin C.C. Johansson; Andy McNally; Ngoc T. Vo (2007). "Enantioselective organocatalysis". Drug Discovery Today. 12 (1/2): 8–27. doi:10.1016/j.drudis.2006.11.004. PMID 17198969.
- ^ Dieter Enders; Christoph Grondal; Matthias R. M. Hüttl (2007). "Asymmetric Organocatalytic Domino Reactions". Angew. Chem. Int. Ed. 46 (10): 1570–1581. doi:10.1002/anie.200603129. PMID 17225236.
- ^ Peter I. Dalko; Lionel Moisan (2001). "Enantioselective Organocatalysis". Angew. Chem. Int. Ed. 40 (20): 3726–3748. doi:10.1002/1521-3773(20011015)40:20<3726::AID-ANIE3726>3.0.CO;2-D. PMID 11668532.
- ^ International Patent WO 2006068611 A1 20060629 " Direct Homogeneous and Heterogeneous Organic Acid and Amino Acid-Catalyzed Modification of Amines and Alcohols" Inventors: Armando Córdova, Stockholm, Sweden; Jonas Hafrén, Stockholm, Sweden.
- ^ Example 4 in U.S. Patent 3,975,440 August 17, 1976, Filed Dec. 9, 1970 Zoltan G. Hajos and David R. Parrish.
- ^ "2021 Nobel Prize in chemistry". Nobel Prize. Retrieved 6 October 2021.
- ^ List, B. (2010). "Emil Knoevenagel and the Roots of Aminocatalysis". Angewandte Chemie International Edition in English. 49 (10): 1730–1734. doi:10.1002/anie.200906900. PMID 20175175.
- ^ Neises, Bernhard; Steglich, Wolfgang (July 1978). "Simple Method for the Esterification of Carboxylic Acids". Angewandte Chemie International Edition in English. 17 (7): 522–524. doi:10.1002/anie.197805221.
- ^ Basavaiah, Deevi; Rao, Anumolu Jaganmohan; Satyanarayana, Tummanapalli (March 2003). "Recent Advances in the Baylis−Hillman Reaction and Applications". Chemical Reviews. 103 (3): 811–892. doi:10.1021/cr010043d. PMID 12630854.
- ^ Stetter, Hermann (1976). "Catalyzed Addition of Aldehydes to Activated Double Bonds—A New Synthetic Approach". Angewandte Chemie International Edition in English. 15 (11): 639–647. doi:10.1002/anie.197606391. ISSN 1521-3773.
- ^ MacMillan, David W. C. (2008). "The advent and development of organocatalysis". Nature. 455 (7211). Springer Science and Business Media LLC: 304–308. Bibcode:2008Natur.455..304M. doi:10.1038/nature07367. ISSN 0028-0836. PMID 18800128. S2CID 205215034.
- ^ Z. G. Hajos, D. R. Parrish, German Patent DE 2102623 1971
- ^ Zoltan G. Hajos; David R. Parrish (1974). "Asymmetric synthesis of bicyclic intermediates of natural product chemistry". J. Org. Chem. 39 (12): 1615–1621. doi:10.1021/jo00925a003.
- ^ R. B. Woodward; E. Logusch; K. P. Nambiar; K. Sakan; D. E. Ward; B. W. Au-Yeung; P. Balaram; L. J. Browne; et al. (1981). "Asymmetric total synthesis of erythromcin. 1. Synthesis of an erythronolide A secoacid derivative via asymmetric induction". J. Am. Chem. Soc. 103 (11): 3210–3213. Bibcode:1981JAChS.103.3210W. doi:10.1021/ja00401a049.
- ^ B. -F. Sun (2015). "Total synthesis of natural and pharmaceutical products powered by organocatalytic reactions". Tetrahedron Lett. 56 (17): 2133–2140. doi:10.1016/j.tetlet.2015.03.046.
- ^ Wang, Zhi-Xian; Tu, Yong; Frohn, Michael; Zhang, Jian-Rong; Shi, Yian (1997-11-01). "An Efficient Catalytic Asymmetric Epoxidation Method". Journal of the American Chemical Society. 119 (46): 11224–11235. Bibcode:1997JAChS.11911224W. doi:10.1021/ja972272g. ISSN 0002-7863.
- ^ Bertelsen, Søren (2009). "Organocatalysis—after the gold rush". Chemical Society Reviews. 38 (8): 2178–89. doi:10.1039/b903816g. PMID 19623342.
- ^ Gaunt, M. J.; Johansson, C. C. C.; McNally, A.; Vo, N. T. (2007). "Enantioselective organocatalysis". Drug Discovery Today. 12 (1–2): 8–27. doi:10.1016/j.drudis.2006.11.004. PMID 17198969.
- ^ Kucherenko, A. S.; Siyutkin, D. E.; Maltsev, O. V.; Kochetkov, S. V.; Zlotin, S. G. (2013). "Asymmetric organocatalysis: From proline to highly efficient immobilized organocatalysts". Russian Chemical Bulletin. 61 (7): 1313. doi:10.1007/s11172-012-0177-4. S2CID 93168492.
- ^ Gérald Lelais; David W. C. MacMillan (2006). "Modern Strategies in Organic Catalysis: The Advent and Development of Iminium Activation" (PDF). Aldrichimica Acta. 39 (3): 79.
- ^ Erkkilä, Anniinä; Majander, Inkeri; Pihko, Petri M. (2007). "Iminium Catalysis". Chem. Rev. 107 (12): 5416–5470. doi:10.1021/cr068388p. PMID 18072802.
- ^ Nis Halland; Tore Hansen; Karl Anker Jørgensen (2003). "Organocatalytic Asymmetric Michael Reaction of Cyclic 1,3-Dicarbonyl Compounds and α,β-Unsaturated Ketones – A Highly Atom-Economic Catalytic One-Step Formation of Optically Active Warfarin Anticoagulant". Angew. Chem. Int. Ed. 42 (40): 4955–4957. doi:10.1002/anie.200352136. PMID 14579449.
- ^ Sandra Lee; David W. C. MacMillan (2007). "Organocatalytic Vinyl and Friedel-Crafts Alkylations with Trifluoroborate Salts" (PDF). J. Am. Chem. Soc. 129 (50): 15438–15439. doi:10.1021/ja0767480. PMID 18031044. S2CID 34848947.
- ^ Madarász, Ádám; Dósa, Zsolt; Varga, Szilárd; Soós, Tibor; Csámpai, Antal; Pápai, Imre (July 2016). "Thiourea Derivatives as Brønsted Acid Organocatalysts" (PDF). ACS Catalysis. 6 (7): 4379–4387. doi:10.1021/acscatal.6b00618.
- ^ Parvin, Tasneem; Yadava, Rahul; Choudhury, Lokman (2020). "Recent applications of thiourea-based organocatalysts in asymmetric multicomponent reactions (AMCRs)". Org. Biomol. Chem. 18 (29): 5513–5532. doi:10.1039/D0OB00595A. PMID 32644077.
External links
[edit] Media related to Organocatalysis at Wikimedia Commons
Media related to Organocatalysis at Wikimedia Commons The dictionary definition of organocatalysis at Wiktionary
The dictionary definition of organocatalysis at Wiktionary Quotations related to Organocatalysis at Wikiquote
Quotations related to Organocatalysis at Wikiquote
Organocatalysis
View on GrokipediaFundamentals
Definition and Principles
Organocatalysis is defined as a type of catalysis that utilizes small organic molecules, typically with molecular weights below 2000 Da and composed mainly of elements such as carbon, hydrogen, nitrogen, oxygen, sulfur, and phosphorus, to accelerate chemical reactions under mild conditions without incorporating metals or enzymes.[3][4][5] This approach distinguishes organocatalysis from biocatalysis, which depends on enzymatic macromolecules for selectivity and efficiency, and from traditional metal catalysis, which relies on transition metal complexes to facilitate transformations.[6][4] The foundational principles of organocatalysis center on the deployment of sub-stoichiometric amounts of these organic catalysts to activate substrates, primarily through covalent or non-covalent interactions that generate reactive intermediates and lower the energy barrier for the reaction.[7][8] In a typical catalytic cycle, the organocatalyst interacts with the substrate to form an activated intermediate, which then undergoes transformation to yield the product while regenerating the catalyst for reuse, as illustrated in the general scheme: substrate + organocatalyst → activated intermediate → product + organocatalyst.[9] These catalysts often operate effectively in the presence of water and air, enabling reactions at ambient temperatures and promoting sustainable synthetic methodologies.[10] Organocatalysis frequently targets enantioselectivity, allowing the production of chiral molecules with high stereochemical control using chiral organic catalysts.[4]Advantages and Comparisons
Organocatalysis offers several key advantages over traditional catalytic methods, primarily due to its reliance on non-metallic, organic small molecules as catalysts. These catalysts are inherently environmentally friendly, as they eliminate the use of toxic transition metals, thereby reducing hazardous waste and contamination risks in product purification.[11] Additionally, organocatalytic reactions typically proceed under mild conditions, such as room temperature and in aqueous or protic media, which minimizes energy consumption and enhances compatibility with sensitive functional groups without requiring anhydrous setups or inert atmospheres.[11] The ease of handling stems from the stability of organocatalysts to air and moisture, allowing straightforward laboratory operations and scalability.[12] Furthermore, their low cost arises from the use of readily available, often commercially sourced organic compounds, making them economically viable for both academic and industrial applications.[13] High selectivity is another hallmark, with many processes achieving enantiomeric excesses exceeding 99% and diastereomeric ratios greater than 20:1, enabling precise control over stereochemistry in complex syntheses.[11] In comparison to transition-metal catalysis, organocatalysis stands out by avoiding metal residues, which is particularly beneficial in pharmaceutical synthesis where trace metals can pose toxicity concerns or complicate regulatory approval.[13] Metal-based methods often demand rigorous exclusion of oxygen and water, along with extensive purification to remove residues, whereas organocatalysts operate robustly in ambient conditions without such constraints. Relative to biocatalysis, organocatalysis provides a broader substrate scope, accommodating non-natural or chemically unstable substrates that enzymes may not tolerate, and facilitates easier scale-up due to the chemical tunability and robustness of organic catalysts without the need for biological expression systems.[14] Compared to stoichiometric reagents, organocatalysis enhances efficiency by enabling turnover, significantly reducing waste generation in transformations like reductions or oxidations, where stoichiometric approaches produce large amounts of byproducts.[15] Quantitative performance metrics underscore these benefits, with organocatalysts often exhibiting high turnover numbers (TON) and frequencies (TOF). These values demonstrate catalytic efficiency comparable to or exceeding some metal systems in selective transformations. Organocatalysis aligns closely with green chemistry principles, such as waste prevention and atom economy, by maximizing substrate incorporation and minimizing auxiliary materials; optimized syntheses of organocatalysts yield low E-factors, reflecting reduced overall waste compared to multi-step metal catalyst preparations. This sustainability is further enhanced through recyclable organocatalysts, which can be reused for multiple cycles without loss of activity, supporting solvent-free or low-solvent processes.[11]Historical Development
Early Discoveries
The origins of organocatalysis trace back to the 19th century, with the discovery of cyanide as a catalyst for the benzoin condensation. In 1832, Justus von Liebig and Friedrich Wöhler reported the cyanide-mediated coupling of two molecules of benzaldehyde to form benzoin, marking one of the earliest documented examples of a small organic molecule facilitating a carbon-carbon bond formation without the need for metals.[16] This reaction, initially observed during investigations into bitter almond oil, demonstrated the potential of cyanide ions (derived from sources like potassium cyanide) to activate aldehydes through nucleophilic addition, though it was not recognized as "organocatalysis" at the time.[16] Early 20th-century advances included Wilhelm Ostwald's 1909 vision of organic catalysts mimicking enzymes and Georg Bredig's 1912 demonstration of enantioselective hydrogenation using cinchona alkaloids, laying foundations for asymmetric organocatalysis.[2] Progress in organocatalysis remained sporadic through the early 20th century until the mid-20th century, when amino acids emerged as effective chiral catalysts for asymmetric synthesis. In the 1970s, independent reports by Eder, Sauer, and Wiechert at Schering AG, as well as Hajos and Parrish at Hoffmann-La Roche, highlighted L-proline as a catalyst for intramolecular aldol reactions.[17] Specifically, these groups demonstrated that L-proline enables the enantioselective cyclization of triketones, such as 2-methyl-1,3-cyclohexanedione with methyl vinyl ketone, to produce bicyclic enediones. This proline-catalyzed aldol reaction achieved high enantiomeric excess, typically around 93% ee for the Wieland-Miescher ketone precursor, establishing it as a benchmark for stereocontrol using a simple organic molecule. These early discoveries laid the groundwork for organocatalysis but faced significant limitations, primarily in substrate scope and applicability beyond natural product synthesis. The reactions were often confined to specific intramolecular contexts or activated substrates, with challenges in extending them to intermolecular processes or diverse functional groups, which restricted broader adoption until later advancements. Proline, functioning as an amine-based catalyst through enamine formation, exemplified this potential but underscored the need for expanded methodologies.[17]Modern Milestones
The year 2000 marked a pivotal surge in organocatalysis, with independent reports from David W. C. MacMillan and Benjamin List demonstrating highly enantioselective reactions using simple organic molecules. MacMillan's group introduced chiral imidazolidinone catalysts derived from amino acids, enabling the first general organocatalytic Diels-Alder reaction between α,β-unsaturated aldehydes and electron-rich alkenes with up to 99% enantiomeric excess (ee). Concurrently, List and colleagues showcased L-proline as an efficient catalyst for the direct intermolecular aldol reaction between unmodified ketones and aldehydes, achieving selectivities up to 76% ee and establishing enamine activation as a foundational mode. These publications, appearing in Journal of the American Chemical Society, catalyzed widespread adoption by highlighting organocatalysis's potential for asymmetric synthesis without metals. Subsequent years saw rapid expansion into new catalyst classes, broadening organocatalysis's scope. In 2004, Eric N. Jacobsen's group developed chiral thiourea catalysts, which activate electrophiles via hydrogen bonding, as demonstrated in the enantioselective acyl-Pictet-Spengler reaction of tryptamine derivatives with up to 92% ee. In 2006, Jacobsen advanced thioureas for the asymmetric nitro-Mannich (aza-Henry) reaction, achieving up to 94% ee. Independently, in 2004, Takahiko Akiyama pioneered chiral phosphoric acids as Brønsted acid organocatalysts; Akiyama's BINOL-derived phosphoric acid facilitated direct Mannich reactions of 1,3-dicarbonyls with imines in up to 83% ee. Concurrently, Mikiji Terada developed analogous catalysts for direct Mannich reactions (Angew. Chem. Int. Ed. 2005, 44, 1684), achieving up to 89% ee, ushering in a versatile class for carbon-carbon bond formation. The field's impact culminated in the 2021 Nobel Prize in Chemistry, awarded jointly to List and MacMillan for their foundational contributions to asymmetric organocatalysis, recognizing its role in enabling precise molecular construction with sustainable, metal-free methods.[18] This accolade underscored organocatalysis's transformation from niche to mainstream, influencing pharmaceutical and materials synthesis. By the 2020s, organocatalysis integrated with emerging technologies, enhancing efficiency and scope. Recent advances include synergistic photocatalysis, where organocatalysts pair with visible-light mediators for radical-mediated asymmetric transformations, such as enantioselective α-alkylation of aldehydes with up to 95% ee. Machine learning has accelerated catalyst design, with models predicting optimal structures for enamine and iminium activations. These developments reflect organocatalysis's evolution toward predictive, hybrid systems. Publication growth illustrates the field's expansion: fewer than 100 papers annually in the 1990s escalated to ~1,500 per year by the 2020s, driven by accessible catalysts and broad applications.[19]Mechanistic Principles
Activation Modes
Organocatalysis activates substrates through covalent and non-covalent mechanisms that either form transient bonds or employ intermolecular forces to enhance reactivity and selectivity. Covalent modes generate reactive intermediates by bonding the catalyst to the substrate, while non-covalent modes stabilize transition states via weaker interactions, often polarizing bonds without covalent linkage. These strategies enable diverse transformations, with bifunctional approaches integrating multiple modes for cooperative effects.[1] Covalent activation prominently features enamine formation, where a secondary amine reacts with an aldehyde or ketone possessing an α-hydrogen to form a nucleophilic enamine intermediate. The process involves nucleophilic addition to the carbonyl, forming a carbinolamine, followed by protonation, dehydration to an iminium ion, and deprotonation from the α-carbon to yield the enamine. The enamine's elevated HOMO energy mimics enolate reactivity, facilitating nucleophilic additions like carbon-carbon bond formation. A general scheme is: More precisely, the iminium is formed as [R^1R^2N=CH(R^3)-CH2-R^4]^+ , then loses α-proton to enamine R^1R^2N-CH(R^3)=CH-R^4. This activation was established in asymmetric contexts by List et al. in 2000, demonstrating high enantioselectivity in aldol reactions.[20] Iminium ion activation represents another covalent strategy, particularly with secondary amines and α,β-unsaturated carbonyls. The secondary amine condenses with the aldehyde to form a carbinolamine, which dehydrates to an electrophilic iminium ion with lowered LUMO energy, enhancing its reactivity as an acceptor in cycloadditions or conjugate additions. The process for a general secondary amine and enal is: Ahrendt, Borths, and MacMillan introduced this mode in 2000 for enantioselective Diels-Alder reactions using chiral imidazolidinone catalysts, achieving up to 99% ee.[21] Non-covalent activation leverages hydrogen bonding, where catalyst donors like thioureas engage acceptor sites on electrophiles, such as carbonyl oxygens, to withdraw electron density and boost reactivity. A typical interaction is: Okino et al. demonstrated this in 2003 using thiourea derivatives, enabling asymmetric Michael additions with 92% ee. Additional non-covalent modes include π-π stacking, where aromatic catalyst-substrate overlaps stabilize oriented transition states, and electrostatic interactions, involving charge-charge or dipole alignments to preorganize reactants. These contribute to rate acceleration and specificity without covalent commitment.[22] Bifunctional catalysis merges activation modes in one scaffold, such as an amine for covalent nucleophile generation paired with a hydrogen-bond donor for electrophile polarization, allowing ternary complex formation for enhanced efficiency. This dual activation, as in enamine-hydrogen bonding synergy, promotes selective bond formation and has driven many high-impact syntheses.Enantioselectivity and Stereocontrol
Enantioselectivity in organocatalysis is achieved when a chiral organic catalyst induces asymmetry in the formation of chiral products by preferentially stabilizing one enantiotopic face of a prochiral substrate over the other, resulting in differential transition state energies that favor one enantiomer. This kinetic resolution of enantiomeric pathways relies on noncovalent interactions such as hydrogen bonding, electrostatic forces, and steric repulsion to create an energy bias, typically on the order of 1-3 kcal/mol, sufficient to produce high enantiomeric excesses (ee). The enantiomeric excess, defined as ee = |(R - S)/(R + S)| × 100% where R and S represent the amounts of each enantiomer, serves as the primary metric for quantifying stereocontrol, with values above 90% considered excellent for synthetic applications.[23] The relationship between this energy difference and observed selectivity is captured by the equation where is the difference in Gibbs free energies of activation for the competing transition states, is the gas constant, and is the absolute temperature; this formulation, derived from the Boltzmann distribution applied to rate constants, underscores how even modest values (e.g., 2 kcal/mol at 298 K) can yield ee >90%. A foundational example of stereocontrol modeling is the Houk-List transition state model for enamine catalysis in proline-mediated aldol reactions, which illustrates facial selectivity through diastereomeric transition states where the enamine derived from proline attacks the aldehyde's Re or Si face. In the preferred pathway, the aldehyde's carbonyl coordinates to the pyrrolidine nitrogen via hydrogen bonding from the carboxylic acid, while steric shielding by the proline ring disfavors the opposite approach, leading to predictable syn aldol products with ee up to 99%. This model, validated computationally, has become a benchmark for rationalizing and designing enantioselective enamine processes. Key factors enhancing enantioselectivity include precise catalyst-substrate matching, where complementary steric bulk and electronic properties—such as the fit between a catalyst's chiral pocket and substrate's reactive site—amplify transition state differentiation; mismatches can reduce ee by 20-50% or more. Solvent choice significantly modulates these interactions, with nonpolar solvents like toluene preserving hydrogen bonds and π-stacking for higher ee (often >95%), whereas protic or highly polar solvents may solvate key interactions, eroding selectivity by up to 30%. Temperature control further refines outcomes, as lowering the reaction temperature (e.g., to -20°C) exaggerates small differences, routinely achieving ee >95% in sensitive systems by favoring kinetic resolution over thermal equilibration. Absolute configurations of products are reliably predicted using computational tools like density functional theory (DFT), which simulate transition state geometries and energies to forecast stereochemical preferences with errors typically <5% ee, enabling catalyst optimization without extensive experimentation. Recent computational studies, as of 2025, highlight the role of solute-solvent van der Waals interactions in fine-tuning selectivity. Hydrogen bonding frequently underpins this stereocontrol across organocatalytic modes.[23][24]Organocatalyst Classes
Amine-Based Catalysts
Amine-based catalysts represent a cornerstone of organocatalysis, primarily enabling covalent activation of carbonyl substrates through the formation of enamines or iminium ions. These catalysts, typically derived from secondary or primary amines, facilitate nucleophilic addition pathways that are essential for asymmetric C-C bond formation. Proline and its derivatives exemplify this class, offering simplicity, biocompatibility, and high stereocontrol due to their rigid pyrrolidine structure and bifunctional nature. L-Proline, a natural α-amino acid, acts as a bifunctional organocatalyst, with its secondary amine group forming reactive intermediates and its carboxylic acid enabling additional interactions. In the Hajos-Parrish-Eder-Sauer-Wiechert reaction, L-proline catalyzes the intramolecular aldol condensation of 2-methyl-1,3-cyclohexanedione with methyl vinyl ketone, producing a chiral bicyclic aldol product in up to 93% enantiomeric excess (ee). This reaction, independently reported by two groups, established proline as a viable asymmetric catalyst for steroid precursor synthesis. The mechanism begins with the condensation of proline's amine with an aldehyde or ketone to form an enamine intermediate: This enamine then acts as a nucleophile in an aldol addition to another carbonyl, followed by hydrolysis to regenerate proline and yield the β-hydroxy carbonyl product. Such processes achieve >90% ee in various intermolecular aldol reactions, highlighting proline's broad scope for enantioselective C-C bond formation. Imidazolidin-4-one catalysts, pioneered by MacMillan in 2000, extend amine-based activation through iminium ion formation, featuring a chiral pyrrolidine ring derived from (S)-proline or similar scaffolds. These catalysts enable highly enantioselective Diels-Alder reactions between α,β-unsaturated aldehydes and electron-rich alkenes, delivering cycloadducts with up to 99% ee and demonstrating iminium catalysis as a metal-free alternative to Lewis acid activation. In Friedel-Crafts alkylations, the same imidazolidinones promote the addition of indoles to enals, affording α-aryl aldehydes in 94% ee, thus expanding the utility of amine catalysts to aromatic substitutions with excellent stereocontrol. Variants of amine-based catalysts include primary amine derivatives of cinchona alkaloids, which support enamine-mediated transformations like asymmetric Michael additions and aldol reactions, often achieving >95% ee due to their rigid quinuclidine framework. Peptide amines, such as tri- or tetrapeptides incorporating proline residues, further diversify this class by providing multidentate activation for conjugate additions, with select examples yielding products in >90% ee for C-C bond formations. These developments underscore the versatility of amine-based catalysts in achieving high enantioselectivity across diverse synthetic scopes.Hydrogen-Bond Donor Catalysts
Hydrogen-bond donor catalysts in organocatalysis operate through non-covalent interactions, primarily by forming hydrogen bonds with electron-deficient substrates to activate them for nucleophilic attack. These catalysts, often derived from urea, thiourea, or squaramide scaffolds, lower the lowest unoccupied molecular orbital (LUMO) energy of electrophiles such as carbonyl compounds, enhancing their reactivity without forming covalent intermediates. This activation mode is particularly effective for asymmetric transformations involving imines, enones, and nitroalkenes, enabling high levels of enantioselectivity in metal-free conditions.[25] Thioureas represent one of the most prominent classes of hydrogen-bond donor catalysts, featuring a core structure of (NH)_2C=S attached to a chiral backbone, such as trans-1,2-diaminocyclohexane, often substituted with electron-withdrawing groups like 3,5-bis(trifluoromethyl)phenyl moieties to enhance acidity. The seminal introduction of chiral thioureas was reported by Vachal and Jacobsen in 2002, who demonstrated their efficacy in the enantioselective addition of silyl ketene acetals to N-Boc aldimines, achieving β-amino acid derivatives with up to 94% enantiomeric excess (ee).[26] In 2004, the same group extended thioureas to the acyl-Pictet-Spengler reaction, where they bind anionic intermediates to promote cyclization of tryptamine derivatives into β-carbolines with ee values exceeding 99%, highlighting their role in anion-binding organocatalysis.[27] The mechanism involves dual hydrogen bonding from the thiourea NH groups to the substrate's electronegative atoms, stabilizing transition states and enforcing stereocontrol through the chiral framework. Squaramides, cyclic four-membered ring analogs of ureas with enhanced hydrogen-bond donor strength due to their conjugated, electron-deficient core, have emerged as superior alternatives for challenging activations. First applied in asymmetric catalysis by Malerich, Hagihara, and Rawal in 2008, chiral squaramides catalyzed the Michael addition of 1,3-dicarbonyl compounds to nitroolefins, delivering products with up to 99% ee and broad substrate scope.[28] Their increased acidity compared to thioureas allows for tighter binding and more effective LUMO lowering, as depicted in the interaction:Catalyst-NH ⋯ O=C-EWG
Catalyst-NH ⋯ O=C-EWG