
Community hub
Recent from talks
Contribute something
Nothing was collected or created yet.
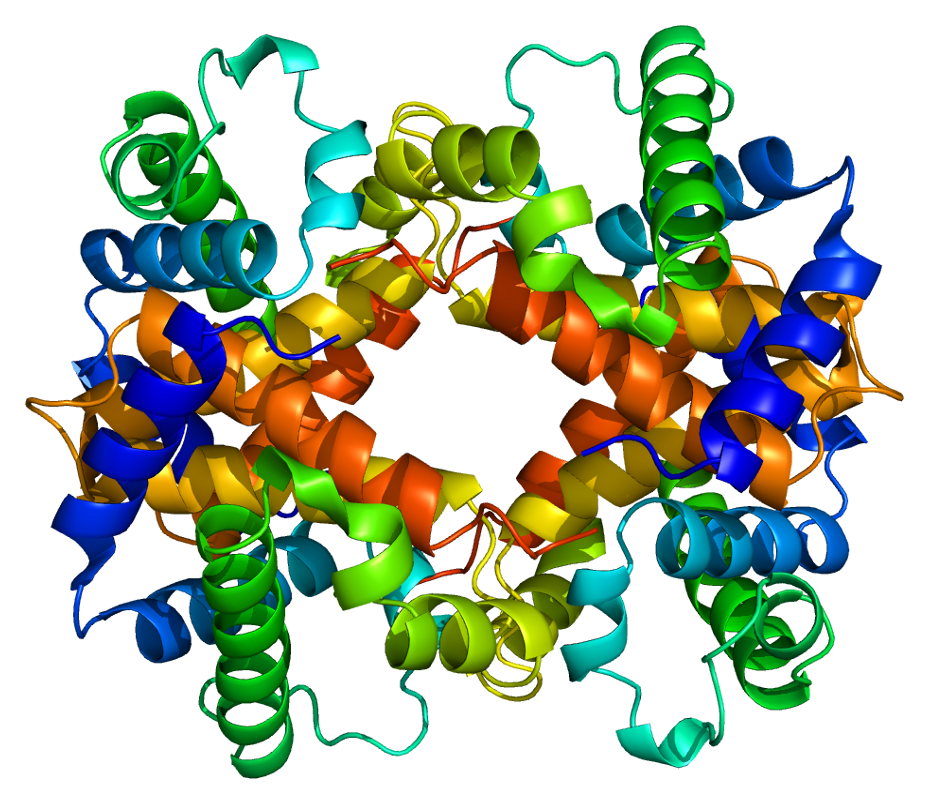
Hemoglobin subunit alpha
View on Wikipedia





Hemoglobin subunit alpha, Hemoglobin, alpha 1,[5] is a hemoglobin protein that in humans is encoded by the HBA1 gene.[6]
Gene
[edit]The human alpha globin gene cluster located on chromosome 16 spans about 30 kb and includes seven loci: 5'- zeta - pseudozeta - mu - pseudoalpha-1 - alpha-2 - alpha-1 - theta - 3'. The alpha-2 (HBA2) and alpha-1 (HBA1; this gene) coding sequences are identical. These genes differ slightly over the 5' untranslated regions and the introns, but they differ significantly over the 3' untranslated regions.[6]
Protein
[edit]Two alpha chains plus two beta chains constitute HbA, which in normal adult life accounts for about 97% of the total hemoglobin; alpha chains combine with delta chains to constitute HbA-2, which with fetal hemoglobin (HbF), composed of alpha and gamma chains, make up the remaining 3% of adult hemoglobin.[6]
Clinical significance
[edit]Alpha thalassemias result from deletions of each of the alpha genes as well as deletions of both HBA2 and HBA1; some nondeletion alpha thalassemias have also been reported.[6]
Interactions
[edit]Hemoglobin subunit alpha has been shown to interact with hemoglobin subunit beta (HBB).[7][8]
See also
[edit]References
[edit]- ^ a b c GRCh38: Ensembl release 89: ENSG00000206172 – Ensembl, May 2017
- ^ a b c GRCm38: Ensembl release 89: ENSMUSG00000069919 – Ensembl, May 2017
- ^ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ "HBA1 gene: MedlinePlus Genetics".
- ^ a b c d "Entrez Gene: HBA1 hemoglobin, alpha 1".
- ^ Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S, Timm J, Mintzlaff S, Abraham C, Bock N, Kietzmann S, Goedde A, Toksöz E, Droege A, Krobitsch S, Korn B, Birchmeier W, Lehrach H, Wanker EE (September 2005). "A human protein-protein interaction network: a resource for annotating the proteome". Cell. 122 (6): 957–68. doi:10.1016/j.cell.2005.08.029. hdl:11858/00-001M-0000-0010-8592-0. PMID 16169070. S2CID 8235923.
- ^ Shaanan B (November 1983). "Structure of human oxyhaemoglobin at 2.1 A resolution". J. Mol. Biol. 171 (1): 31–59. doi:10.1016/S0022-2836(83)80313-1. PMID 6644819.
Further reading
[edit]- Turbpaiboon C, Svasti S, Sawangareetakul P, et al. (2002). "Hb Siam [alpha15(A13)Gly→Arg (alpha1) (GGT→CGT)] is a typical alpha chain hemoglobinopathy without an alpha-thalassemic effect". Hemoglobin. 26 (1): 77–81. doi:10.1081/HEM-120002944. PMID 11939517. S2CID 85035930.
- Yalçin A, Avcu F, Beyan C, et al. (1995). "A case of HB J-Meerut (or Hb J-Birmingham) [alpha 120(H3)Ala→Glu]". Hemoglobin. 18 (6): 433–5. doi:10.3109/03630269409045775. PMID 7713747.
- Giardina B, Messana I, Scatena R, Castagnola M (1995). "The multiple functions of hemoglobin". Crit. Rev. Biochem. Mol. Biol. 30 (3): 165–96. doi:10.3109/10409239509085142. PMID 7555018.
- Higgs DR, Vickers MA, Wilkie AO, et al. (1989). "A review of the molecular genetics of the human alpha-globin gene cluster". Blood. 73 (5): 1081–104. doi:10.1182/blood.V73.5.1081.1081. PMID 2649166.
- Schillirò G, Russo-Mancuso G, Dibenedetto SP, et al. (1992). "Six rare hemoglobin variants found in Sicily". Hemoglobin. 15 (5): 431–7. doi:10.3109/03630269108998862. PMID 1802885.
- Vafa M, Troye-Blomberg M, Anchang J, et al. (2008). "Multiplicity of Plasmodium falciparum infection in asymptomatic children in Senegal: relation to transmission, age and erythrocyte variants". Malar. J. 7: 17. doi:10.1186/1475-2875-7-17. PMC 2267475. PMID 18215251.
- Datta P, Chakrabarty S, Chakrabarty A, Chakrabarti A (2008). "Membrane interactions of hemoglobin variants, HbA, HbE, HbF and globin subunits of HbA: effects of aminophospholipids and cholesterol". Biochim. Biophys. Acta. 1778 (1): 1–9. doi:10.1016/j.bbamem.2007.08.019. PMID 17916326.
- Taylor JG, Ackah D, Cobb C, et al. (2008). "Mutations and polymorphisms in hemoglobin genes and the risk of pulmonary hypertension and death in sickle cell disease". Am. J. Hematol. 83 (1): 6–14. doi:10.1002/ajh.21035. PMC 3509176. PMID 17724704.
- Sahu SC, Simplaceanu V, Gong Q, et al. (2007). "Insights into the solution structure of human deoxyhemoglobin in the absence and presence of an allosteric effector". Biochemistry. 46 (35): 9973–80. doi:10.1021/bi700935z. PMC 2532491. PMID 17691822.
- Sorour Y, Heppinstall S, Porter N, et al. (2007). "Is routine molecular screening for common alpha-thalassaemia deletions necessary as part of an antenatal screening programme?". Journal of Medical Screening. 14 (2): 60–1. doi:10.1258/096914107781261981. PMID 17626702. S2CID 24823660.
- Hung CC, Lee CN, Chen CP, et al. (2007). "Molecular assay of -alpha(3.7) and -alpha(4.2) deletions causing alpha-thalassemia by denaturing high-performance liquid chromatography". Clin. Biochem. 40 (11): 817–21. doi:10.1016/j.clinbiochem.2007.03.018. PMID 17512924.
- Ye BC, Zhang Z, Lei Z (2007). "Molecular analysis of alpha/beta-thalassemia in a southern Chinese population". Genet. Test. 11 (1): 75–83. doi:10.1089/gte.2006.0502. PMID 17394396.
- Dilley J, Ganesan A, Deepa R, et al. (2007). "Association of A1C with cardiovascular disease and metabolic syndrome in Asian Indians with normal glucose tolerance". Diabetes Care. 30 (6): 1527–32. doi:10.2337/dc06-2414. PMID 17351274.
- Fonseka PV, Vasudevan G, Clarizia LJ, McDonald MJ (2007). "Temperature dependent soret spectral band shifts accompany human CN-mesohemoglobin assembly". Protein J. 26 (4): 257–63. doi:10.1007/s10930-006-9067-7. PMID 17191128. S2CID 24601675.
- Sankar VH, Arya V, Tewari D, et al. (2007). "Genotyping of alpha-thalassemia in microcytic hypochromic anemia patients from North India". J. Appl. Genet. 47 (4): 391–5. doi:10.1007/BF03194650. PMID 17132905. S2CID 19194610.
- Origa R, Sollaino MC, Giagu N, et al. (2007). "Clinical and molecular analysis of haemoglobin H disease in Sardinia: haematological, obstetric and cardiac aspects in patients with different genotypes". Br. J. Haematol. 136 (2): 326–32. doi:10.1111/j.1365-2141.2006.06423.x. PMID 17129226. S2CID 23265827.
- Hussein OA, Gefen Y, Zidan JM, et al. (2007). "LDL oxidation is associated with increased blood hemoglobin A1c levels in diabetic patients". Clin. Chim. Acta. 377 (1–2): 114–8. doi:10.1016/j.cca.2006.09.002. PMID 17070510.
- Pan W, Galkin O, Filobelo L, et al. (2007). "Metastable mesoscopic clusters in solutions of sickle-cell hemoglobin". Biophys. J. 92 (1): 267–77. Bibcode:2007BpJ....92..267P. doi:10.1529/biophysj.106.094854. PMC 1697867. PMID 17040989.
- Pistrosch F, Koehler C, Wildbrett J, Hanefeld M (2006). "Relationship between diurnal glucose levels and HbA1c in type 2 diabetes". Horm. Metab. Res. 38 (7): 455–9. doi:10.1055/s-2006-947838. PMID 16933182. S2CID 260166156.
- Chong YM, Tan JA, Zubaidah Z, et al. (2006). "Screening of concurrent alpha-thalassaemia 1 in beta-thalassaemia carriers". Med. J. Malaysia. 61 (2): 217–20. PMID 16898315.
External links
[edit]- GeneReviews/NCBI/NIH/UW entry on Alpha-Thalassemia
- OMIM entries on Alpha-Thalassemia
- Overview of all the structural information available in the PDB for UniProt: P69905 (Hemoglobin subunit alpha) at the PDBe-KB.
PDB gallery | |
|---|---|
|

























































































![1rvw: R STATE HUMAN HEMOGLOBIN [ALPHA V96W], CARBONMONOXY](https://en.wikipedia.org/wiki/File:PDB_1rvw_EBI.jpg)






![1vwt: T STATE HUMAN HEMOGLOBIN [ALPHA V96W], ALPHA AQUOMET, BETA DEOXY](https://en.wikipedia.org/wiki/File:PDB_1vwt_EBI.jpg)














































































