Recent from talks
Metalloprotein
Knowledge base stats:
Talk channels stats:
Members stats:
Metalloprotein
Metalloprotein is a generic term for a protein that contains a metal ion cofactor. A large proportion of all proteins are part of this category. For instance, at least 1000 human proteins (out of ~20,000) contain zinc-binding protein domains although there may be up to 3000 human zinc metalloproteins.
It is estimated that approximately half of all proteins contain a metal. In another estimate, about one quarter to one third of all proteins are proposed to require metals to carry out their functions. Thus, metalloproteins have many different functions in cells, such as storage and transport of proteins, enzymes and signal transduction proteins, or infectious diseases.
Most metals in the human body are bound to proteins. For instance, the relatively high concentration of iron in the human body is mostly due to the iron in hemoglobin.
In metalloproteins, metal ions are usually coordinated by nitrogen, oxygen or sulfur centers belonging to amino acid residues of the protein. These donor groups are often provided by side-chains on the amino acid residues. Especially important are the imidazole substituent in histidine residues, thiolate substituents in cysteine residues, and carboxylate groups provided by aspartate. Given the diversity of the metalloproteome, virtually all amino acid residues have been shown to bind metal centers. The peptide backbone also provides donor groups; these include deprotonated amides and the amide carbonyl oxygen centers. Lead(II) binding in natural and artificial proteins has been reviewed.
In addition to donor groups that are provided by amino acid residues, many organic cofactors function as ligands. Perhaps most famous are the tetradentate N4 macrocyclic ligands incorporated into the heme protein. Inorganic ligands such as sulfide and oxide are also common.
These are the second stage product of protein hydrolysis obtained by treatment with slightly stronger acids and alkalies.
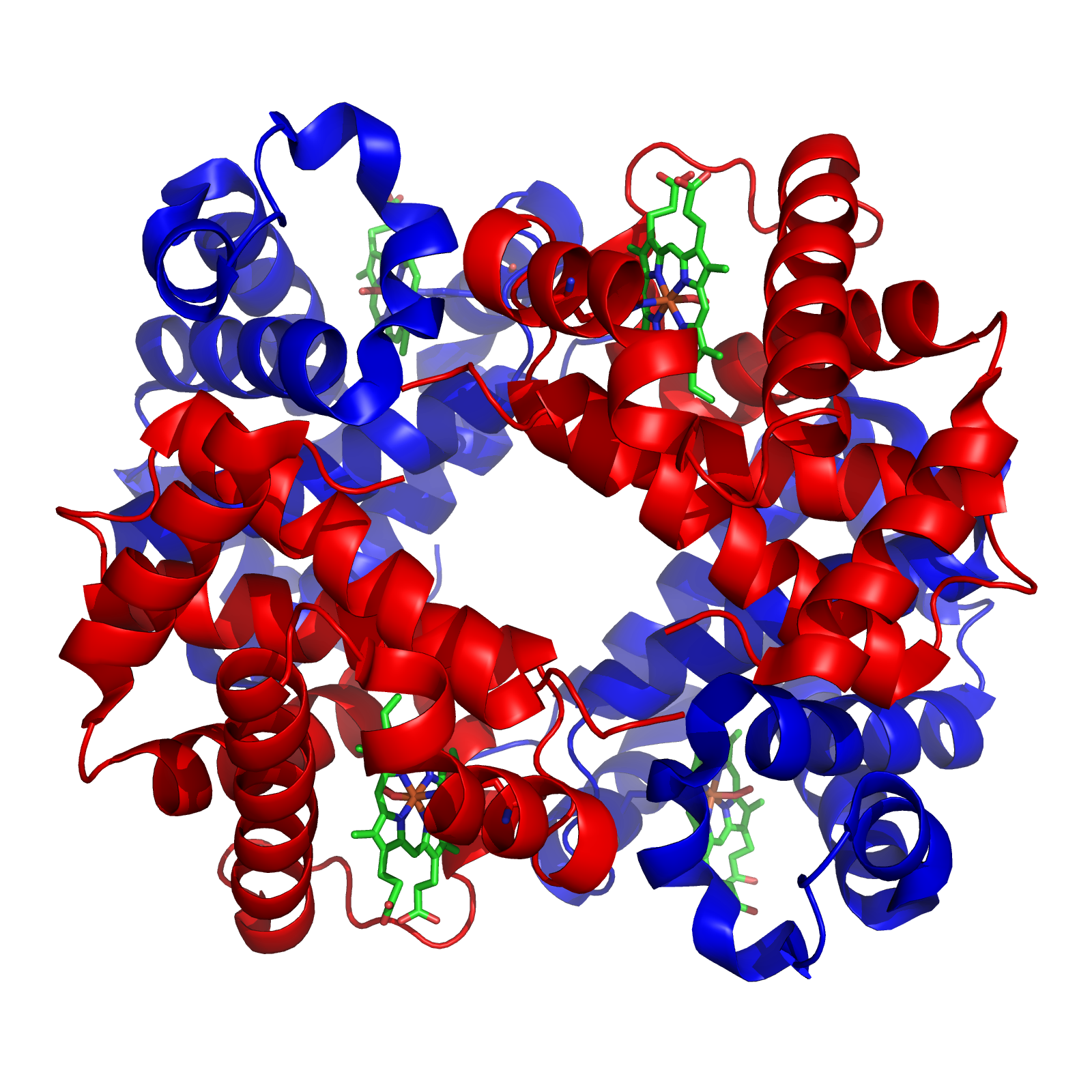
Hemoglobin, which is the principal oxygen-carrier in humans, has four subunits in which the iron(II) ion is coordinated by the planar macrocyclic ligand protoporphyrin IX (PIX) and the imidazole nitrogen atom of a histidine residue. The sixth coordination site contains a water molecule or a dioxygen molecule. By contrast the protein myoglobin, found in muscle cells, has only one such unit. The active site is located in a hydrophobic pocket. This is important as without it the iron(II) would be irreversibly oxidized to iron(III). The equilibrium constant for the formation of HbO2 is such that oxygen is taken up or released depending on the partial pressure of oxygen in the lungs or in muscle. In hemoglobin the four subunits show a cooperativity effect that allows for easy oxygen transfer from hemoglobin to myoglobin.
In both hemoglobin and myoglobin it is sometimes incorrectly stated that the oxygenated species contains iron(III). It is now known that the diamagnetic nature of these species is because the iron(II) atom is in the low-spin state. In oxyhemoglobin the iron atom is located in the plane of the porphyrin ring, but in the paramagnetic deoxyhemoglobin the iron atom lies above the plane of the ring. This change in spin state is a cooperative effect due to the higher crystal field splitting and smaller ionic radius of Fe2+ in the oxyhemoglobin moiety.
Hub AI
Metalloprotein AI simulator
(@Metalloprotein_simulator)
Metalloprotein
Metalloprotein is a generic term for a protein that contains a metal ion cofactor. A large proportion of all proteins are part of this category. For instance, at least 1000 human proteins (out of ~20,000) contain zinc-binding protein domains although there may be up to 3000 human zinc metalloproteins.
It is estimated that approximately half of all proteins contain a metal. In another estimate, about one quarter to one third of all proteins are proposed to require metals to carry out their functions. Thus, metalloproteins have many different functions in cells, such as storage and transport of proteins, enzymes and signal transduction proteins, or infectious diseases.
Most metals in the human body are bound to proteins. For instance, the relatively high concentration of iron in the human body is mostly due to the iron in hemoglobin.
In metalloproteins, metal ions are usually coordinated by nitrogen, oxygen or sulfur centers belonging to amino acid residues of the protein. These donor groups are often provided by side-chains on the amino acid residues. Especially important are the imidazole substituent in histidine residues, thiolate substituents in cysteine residues, and carboxylate groups provided by aspartate. Given the diversity of the metalloproteome, virtually all amino acid residues have been shown to bind metal centers. The peptide backbone also provides donor groups; these include deprotonated amides and the amide carbonyl oxygen centers. Lead(II) binding in natural and artificial proteins has been reviewed.
In addition to donor groups that are provided by amino acid residues, many organic cofactors function as ligands. Perhaps most famous are the tetradentate N4 macrocyclic ligands incorporated into the heme protein. Inorganic ligands such as sulfide and oxide are also common.
These are the second stage product of protein hydrolysis obtained by treatment with slightly stronger acids and alkalies.
Hemoglobin, which is the principal oxygen-carrier in humans, has four subunits in which the iron(II) ion is coordinated by the planar macrocyclic ligand protoporphyrin IX (PIX) and the imidazole nitrogen atom of a histidine residue. The sixth coordination site contains a water molecule or a dioxygen molecule. By contrast the protein myoglobin, found in muscle cells, has only one such unit. The active site is located in a hydrophobic pocket. This is important as without it the iron(II) would be irreversibly oxidized to iron(III). The equilibrium constant for the formation of HbO2 is such that oxygen is taken up or released depending on the partial pressure of oxygen in the lungs or in muscle. In hemoglobin the four subunits show a cooperativity effect that allows for easy oxygen transfer from hemoglobin to myoglobin.
In both hemoglobin and myoglobin it is sometimes incorrectly stated that the oxygenated species contains iron(III). It is now known that the diamagnetic nature of these species is because the iron(II) atom is in the low-spin state. In oxyhemoglobin the iron atom is located in the plane of the porphyrin ring, but in the paramagnetic deoxyhemoglobin the iron atom lies above the plane of the ring. This change in spin state is a cooperative effect due to the higher crystal field splitting and smaller ionic radius of Fe2+ in the oxyhemoglobin moiety.
Recent media