
Community hub
Recent from talks
Contribute something to knowledge base
Content stats: 0 posts, 0 articles, 1 media, 0 notes
Members stats: 0 subscribers, 0 contributors, 0 moderators, 0 supporters
Subscribers
Supporters
Contributors
Moderators
Hub AI
Point mutation AI simulator
(@Point mutation_simulator)
Hub AI
Point mutation AI simulator
(@Point mutation_simulator)
Point mutation
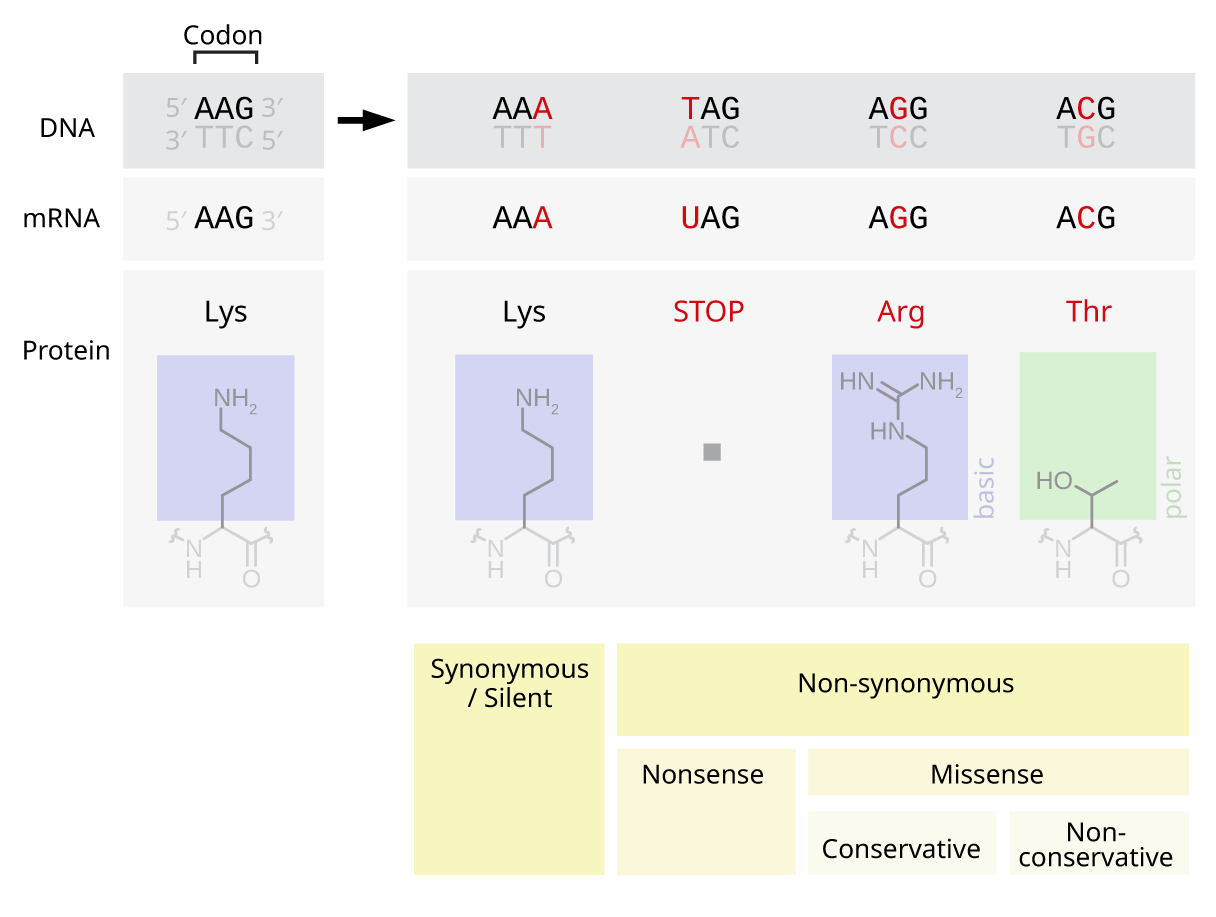
A point mutation is a genetic mutation where a single nucleotide base is changed, inserted or deleted from a DNA or RNA sequence of an organism's genome. Point mutations have a variety of effects on the downstream protein product—consequences that are moderately predictable based upon the specifics of the mutation. These consequences can range from no effect (e.g. synonymous mutations) to deleterious effects (e.g. frameshift mutations), with regard to protein production, composition, and function.
Point mutations usually take place during DNA replication. DNA replication occurs when one double-stranded DNA molecule creates two single strands of DNA, each of which is a template for the creation of the complementary strand. A single point mutation can change the whole DNA sequence. Changing one purine or pyrimidine may change the amino acid that the nucleotides code for.
Point mutations may arise from spontaneous mutations that occur during DNA replication. The rate of mutation may be increased by mutagens. Mutagens can be physical, such as radiation from UV rays, X-rays or extreme heat, or chemical (molecules that misplace base pairs or disrupt the helical shape of DNA). Mutagens associated with cancers are often studied to learn about cancer and its prevention.
There are multiple ways for point mutations to occur. First, ultraviolet (UV) light and higher-frequency light have ionizing capability, which in turn can affect DNA. Reactive oxygen molecules with free radicals, which are a byproduct of cellular metabolism, can also be very harmful to DNA. These reactants can lead to both single-stranded and double-stranded DNA breaks. Third, bonds in DNA eventually degrade, which creates another problem to keep the integrity of DNA to a high standard. There can also be replication errors that lead to substitution, insertion, or deletion mutations.
In 1959 Ernst Freese coined the terms "transitions" or "transversions" to categorize different types of point mutations. Transitions are replacement of a purine base with another purine or replacement of a pyrimidine with another pyrimidine. Transversions are replacement of a purine with a pyrimidine or vice versa. There is a systematic difference in mutation rates for transitions (Alpha) and transversions (Beta). Transition mutations are about ten times more common than transversions.
Nonsense mutations include stop-gain and start-loss. Stop-gain is a mutation that results in a premature termination codon (a stop was gained), which signals the end of translation. This interruption causes the protein to be abnormally shortened. The number of amino acids lost mediates the impact on the protein's functionality and whether it will function whatsoever. Stop-loss is a mutation in the original termination codon (a stop was lost), resulting in abnormal extension of a protein's carboxyl terminus. Start-gain creates an AUG start codon upstream of the original start site. If the new AUG is near the original start site, in-frame within the processed transcript and downstream to a ribosomal binding site, it can be used to initiate translation. The likely effect is additional amino acids added to the amino terminus of the original protein. Frame-shift mutations are also possible in start-gain mutations, but typically do not affect translation of the original protein. Start-loss is a point mutation in a transcript's AUG start codon, resulting in the reduction or elimination of protein production.
Missense mutations code for a different amino acid. A missense mutation changes a codon so that a different protein is created, a non-synonymous change. Conservative mutations result in an amino acid change. However, the properties of the amino acid remain the same (e.g., hydrophobic, hydrophilic, etc.). At times, a change to one amino acid in the protein is not detrimental to the organism as a whole. Most proteins can withstand one or two point mutations before their function changes. Non-conservative mutations result in an amino acid change that has different properties than the wild type. The protein may lose its function, which can result in a disease in the organism. For example, sickle-cell disease is caused by a single point mutation (a missense mutation) in the beta-hemoglobin gene that converts a GAG codon into GUG, which encodes the amino acid valine rather than glutamic acid. The protein may also exhibit a "gain of function" or become activated, such is the case with the mutation changing a valine to glutamic acid in the BRAF gene; this leads to an activation of the RAF protein which causes unlimited proliferative signalling in cancer cells. These are both examples of a non-conservative (missense) mutation.
Silent mutations code for the same amino acid (a "synonymous substitution"). A silent mutation does not affect the functioning of the protein. A single nucleotide can change, but the new codon specifies the same amino acid, resulting in an unmutated protein. This type of change is called synonymous change since the old and new codon code for the same amino acid. This is possible because 64 codons specify only 20 amino acids. Different codons can lead to differential protein expression levels, however.
Point mutation
A point mutation is a genetic mutation where a single nucleotide base is changed, inserted or deleted from a DNA or RNA sequence of an organism's genome. Point mutations have a variety of effects on the downstream protein product—consequences that are moderately predictable based upon the specifics of the mutation. These consequences can range from no effect (e.g. synonymous mutations) to deleterious effects (e.g. frameshift mutations), with regard to protein production, composition, and function.
Point mutations usually take place during DNA replication. DNA replication occurs when one double-stranded DNA molecule creates two single strands of DNA, each of which is a template for the creation of the complementary strand. A single point mutation can change the whole DNA sequence. Changing one purine or pyrimidine may change the amino acid that the nucleotides code for.
Point mutations may arise from spontaneous mutations that occur during DNA replication. The rate of mutation may be increased by mutagens. Mutagens can be physical, such as radiation from UV rays, X-rays or extreme heat, or chemical (molecules that misplace base pairs or disrupt the helical shape of DNA). Mutagens associated with cancers are often studied to learn about cancer and its prevention.
There are multiple ways for point mutations to occur. First, ultraviolet (UV) light and higher-frequency light have ionizing capability, which in turn can affect DNA. Reactive oxygen molecules with free radicals, which are a byproduct of cellular metabolism, can also be very harmful to DNA. These reactants can lead to both single-stranded and double-stranded DNA breaks. Third, bonds in DNA eventually degrade, which creates another problem to keep the integrity of DNA to a high standard. There can also be replication errors that lead to substitution, insertion, or deletion mutations.
In 1959 Ernst Freese coined the terms "transitions" or "transversions" to categorize different types of point mutations. Transitions are replacement of a purine base with another purine or replacement of a pyrimidine with another pyrimidine. Transversions are replacement of a purine with a pyrimidine or vice versa. There is a systematic difference in mutation rates for transitions (Alpha) and transversions (Beta). Transition mutations are about ten times more common than transversions.
Nonsense mutations include stop-gain and start-loss. Stop-gain is a mutation that results in a premature termination codon (a stop was gained), which signals the end of translation. This interruption causes the protein to be abnormally shortened. The number of amino acids lost mediates the impact on the protein's functionality and whether it will function whatsoever. Stop-loss is a mutation in the original termination codon (a stop was lost), resulting in abnormal extension of a protein's carboxyl terminus. Start-gain creates an AUG start codon upstream of the original start site. If the new AUG is near the original start site, in-frame within the processed transcript and downstream to a ribosomal binding site, it can be used to initiate translation. The likely effect is additional amino acids added to the amino terminus of the original protein. Frame-shift mutations are also possible in start-gain mutations, but typically do not affect translation of the original protein. Start-loss is a point mutation in a transcript's AUG start codon, resulting in the reduction or elimination of protein production.
Missense mutations code for a different amino acid. A missense mutation changes a codon so that a different protein is created, a non-synonymous change. Conservative mutations result in an amino acid change. However, the properties of the amino acid remain the same (e.g., hydrophobic, hydrophilic, etc.). At times, a change to one amino acid in the protein is not detrimental to the organism as a whole. Most proteins can withstand one or two point mutations before their function changes. Non-conservative mutations result in an amino acid change that has different properties than the wild type. The protein may lose its function, which can result in a disease in the organism. For example, sickle-cell disease is caused by a single point mutation (a missense mutation) in the beta-hemoglobin gene that converts a GAG codon into GUG, which encodes the amino acid valine rather than glutamic acid. The protein may also exhibit a "gain of function" or become activated, such is the case with the mutation changing a valine to glutamic acid in the BRAF gene; this leads to an activation of the RAF protein which causes unlimited proliferative signalling in cancer cells. These are both examples of a non-conservative (missense) mutation.
Silent mutations code for the same amino acid (a "synonymous substitution"). A silent mutation does not affect the functioning of the protein. A single nucleotide can change, but the new codon specifies the same amino acid, resulting in an unmutated protein. This type of change is called synonymous change since the old and new codon code for the same amino acid. This is possible because 64 codons specify only 20 amino acids. Different codons can lead to differential protein expression levels, however.
Recent media
Recent media