Community hub
Cyanohydrin
View on Wikipedia
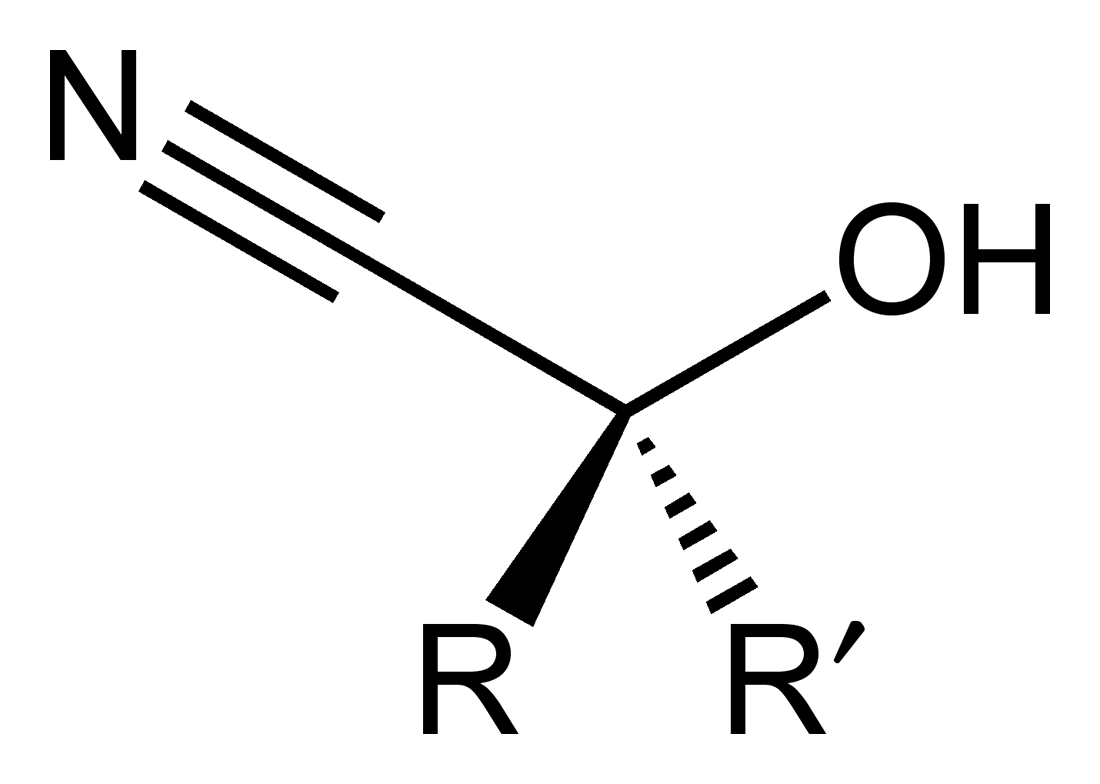
In organic chemistry, a cyanohydrin or hydroxynitrile is a functional group found in organic compounds in which a cyano and a hydroxy group are attached to the same carbon atom. The general formula is R2C(OH)CN, where R is H, alkyl, or aryl. Cyanohydrins are industrially important precursors to carboxylic acids and some amino acids. Cyanohydrins can be formed by the cyanohydrin reaction, which involves treating a ketone or an aldehyde with hydrogen cyanide (HCN) in the presence of excess amounts of sodium cyanide (NaCN) as a catalyst:[1]
- RR’C=O + HCN → RR’C(OH)CN
In this reaction, the nucleophilic CN− ion attacks the electrophilic carbonyl carbon in the ketone, followed by protonation by HCN, thereby regenerating the cyanide anion. Cyanohydrins are also prepared by displacement of sulfite by cyanide salts:[2]

Cyanohydrins are intermediates in the Strecker amino acid synthesis. In aqueous acid, they are hydrolyzed to the α-hydroxy acid.
Preparative methods
[edit]Cyanohydrins are traditionally prepared by the addition of HCN to the corresponding carbonyl. The reaction is typically catalyzed by base or an enzyme.[3][4] Because of the hazards with HCN, other less dangerous cyanation reagents are often used.[5]
- Trimethylsilyl cyanide, affording the silyl ether derivative of the cyanohydrin
- Diethylaluminium cyanide, especially for less reactive carbonyls
- Diethyl phosphorocyanidate (DEPC) and lithium cyanide
- Acyl cyanides (RC(O)CN)

Transhydrocyanation
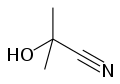
[edit]Acetone cyanohydrin, (CH3)2C(OH)CN is the cyanohydrin of acetone. It is generated as an intermediate in the industrial production of methyl methacrylate.[6] In the laboratory, this liquid serves as a source of HCN. The process is called transhydrocyanation, where acetone cyanohydrin, is used as a source of HCN.[3][4][7] Thus, acetone cyanohydrin can be used for the preparation of other cyanohydrins, for the transformation of HCN to Michael acceptors, and for the formylation of arenes. Treatment of this cyanohydrin with lithium hydride affords anhydrous lithium cyanide:

Asymmetric cyanohydrin formation
[edit]Formation of cyanohydrins introduces a chiral center for aldehydes and for unsymmetrical ketones. The enantioselective hydrocyanation has attracted some attention for the preparation of 2-chloromandelic acid, a drug precursor.[3]
- Some Prominent Cyanohydrins
-
![Glycolonitrile, also called hydroxyacetonitrile or formaldehyde cyanohydrin, is the simplest cyanohydrin.[8]](//upload.wikimedia.org/wikipedia/commons/thumb/4/49/Glyconitrile_Structural_FormulaV1.svg/120px-Glyconitrile_Structural_FormulaV1.svg.png) Glycolonitrile, also called hydroxyacetonitrile or formaldehyde cyanohydrin, is the simplest cyanohydrin.[8]
Glycolonitrile, also called hydroxyacetonitrile or formaldehyde cyanohydrin, is the simplest cyanohydrin.[8] -
-
![Mandelonitrile, occurs in small amounts in the pits of some fruits.[2]](//upload.wikimedia.org/wikipedia/commons/thumb/d/d2/Mandelonitrile-2D-skeletal.svg/120px-Mandelonitrile-2D-skeletal.svg.png) Mandelonitrile, occurs in small amounts in the pits of some fruits.[2]
Mandelonitrile, occurs in small amounts in the pits of some fruits.[2] -
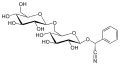 Amygdalin, a naturally occurring cyanogenic glycoside
Amygdalin, a naturally occurring cyanogenic glycoside
![Glycolonitrile, also called hydroxyacetonitrile or formaldehyde cyanohydrin, is the simplest cyanohydrin.[8]](https://en.wikipedia.org/wiki/File:Glyconitrile_Structural_FormulaV1.svg)

![Mandelonitrile, occurs in small amounts in the pits of some fruits.[2]](https://en.wikipedia.org/wiki/File:Mandelonitrile-2D-skeletal.svg)

See also
[edit]References
[edit]- ^ David T. Mowry (1948). "The Preparation of Nitriles". Chem. Rev. 42 (2): 189–283. doi:10.1021/cr60132a001. PMID 18914000.
- ^ a b Corson, B. B.; Dodge, R. A.; Harris, S. A.; Yeaw, J. S. (1941). "Mandelic Acid". Organic Syntheses; Collected Volumes, vol. 1, p. 336.
- ^ a b c North, Michael; Usanov, Dmitry L.; Young, Carl (2008). "Lewis Acid Catalyzed Asymmetric Cyanohydrin Synthesis". Chemical Reviews. 108 (12): 5146–5226. doi:10.1021/cr800255k. PMID 19067648.
- ^ a b Gregory, Robert J. H. (1999). "Cyanohydrins in Nature and the Laboratory: Biology, Preparations, and Synthetic Applications". Chemical Reviews. 99 (12): 3649–3682. doi:10.1021/cr9902906. PMID 11849033.
- ^ Juhl, Martin; Petersen, Allan R.; Lee, Ji-Woong (2021). "CO2-Enabled Cyanohydrin Synthesis and Facile Iterative Homologation Reactions*". Chemistry – A European Journal. 27: 228–232. doi:10.1002/chem.202003623.
- ^ William Bauer, Jr. "Methacrylic Acid and Derivatives" in Ullmann's Encyclopedia of Industrial Chemistry 2002, Wiley-VCH, Weinheim. doi:10.1002/14356007.a16_441. Article Online Posting Date: June 15, 2000
- ^ Haroutounian, S. A. "Acetone Cyanohydrin" Encyclopedia of Reagents for Organic Synthesis 2001, John Wiley & Sons. doi:10.1002/047084289X.ra014
- ^ Gaudry, R. (1955). "Glycolonitrile". Organic Syntheses; Collected Volumes, vol. 3, p. 436.