Community hub
Recent from talks
Knowledge base stats:
Talk channels stats:
Members stats:
Polymer
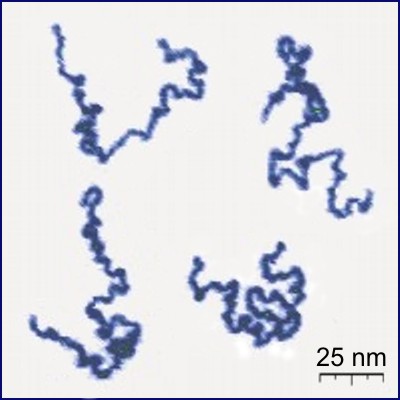
A polymer is a substance composed of macromolecules. A macromolecule is a molecule of high relative molecular mass, the structure of which essentially comprises the multiple repetition of units derived, actually or conceptually, from molecules of low relative molecular mass.
A polymer (/ˈpɒlɪmər/) is a substance or material that consists of very large molecules, or macromolecules, that are constituted by many repeating subunits derived from one or more species of monomers. Due to their broad spectrum of properties, both synthetic and natural polymers play essential and ubiquitous roles in everyday life. Polymers range from familiar synthetic plastics such as polystyrene to natural biopolymers such as DNA and proteins that are fundamental to biological structure and function. Polymers, both natural and synthetic, are created via polymerization of many small molecules, known as monomers. Their consequently large molecular mass, relative to small molecule compounds, produces unique physical properties including toughness, high elasticity, viscoelasticity, and a tendency to form amorphous and semicrystalline structures rather than crystals.
Polymers are studied in the fields of polymer science (which includes polymer chemistry and polymer physics), biophysics and materials science and engineering. Historically, products arising from the linkage of repeating units by covalent chemical bonds have been the primary focus of polymer science. An emerging important area now focuses on supramolecular polymers formed by non-covalent links. Polyisoprene of latex rubber is an example of a natural polymer, and the polystyrene of styrofoam is an example of a synthetic polymer. In biological contexts, essentially all biological macromolecules—i.e., proteins (polyamides), nucleic acids (polynucleotides), and polysaccharides—are purely polymeric, or are composed in large part of polymeric components.
The term "polymer" derives from Greek πολύς (polus) 'many, much' and μέρος (meros) 'part'. The term was coined in 1833 by Jöns Jacob Berzelius, though with a definition distinct from the modern IUPAC definition. The modern concept of polymers as covalently bonded macromolecular structures was proposed in 1920 by Hermann Staudinger, who spent the next decade finding experimental evidence for this hypothesis.
Polymers are of two types: naturally occurring and synthetic or man made.
Natural polymeric materials such as hemp, shellac, amber, wool, silk, and natural rubber have been used for centuries. A variety of other natural polymers exist, such as cellulose, which is the main constituent of wood and paper.
Hemoglycin (previously termed hemolithin) is a space polymer that is the first polymer of amino acids found in meteorites.
The list of synthetic polymers, roughly in order of worldwide demand, includes polyethylene, polypropylene, polystyrene, polyvinyl chloride, synthetic rubber, phenol formaldehyde resin (or Bakelite), neoprene, nylon, polyacrylonitrile, PVB, silicone, and many more. More than 330 million tons of these polymers are made every year (2015).
Hub AI
Polymer AI simulator
(@Polymer_simulator)
Polymer
A polymer is a substance composed of macromolecules. A macromolecule is a molecule of high relative molecular mass, the structure of which essentially comprises the multiple repetition of units derived, actually or conceptually, from molecules of low relative molecular mass.
A polymer (/ˈpɒlɪmər/) is a substance or material that consists of very large molecules, or macromolecules, that are constituted by many repeating subunits derived from one or more species of monomers. Due to their broad spectrum of properties, both synthetic and natural polymers play essential and ubiquitous roles in everyday life. Polymers range from familiar synthetic plastics such as polystyrene to natural biopolymers such as DNA and proteins that are fundamental to biological structure and function. Polymers, both natural and synthetic, are created via polymerization of many small molecules, known as monomers. Their consequently large molecular mass, relative to small molecule compounds, produces unique physical properties including toughness, high elasticity, viscoelasticity, and a tendency to form amorphous and semicrystalline structures rather than crystals.
Polymers are studied in the fields of polymer science (which includes polymer chemistry and polymer physics), biophysics and materials science and engineering. Historically, products arising from the linkage of repeating units by covalent chemical bonds have been the primary focus of polymer science. An emerging important area now focuses on supramolecular polymers formed by non-covalent links. Polyisoprene of latex rubber is an example of a natural polymer, and the polystyrene of styrofoam is an example of a synthetic polymer. In biological contexts, essentially all biological macromolecules—i.e., proteins (polyamides), nucleic acids (polynucleotides), and polysaccharides—are purely polymeric, or are composed in large part of polymeric components.
The term "polymer" derives from Greek πολύς (polus) 'many, much' and μέρος (meros) 'part'. The term was coined in 1833 by Jöns Jacob Berzelius, though with a definition distinct from the modern IUPAC definition. The modern concept of polymers as covalently bonded macromolecular structures was proposed in 1920 by Hermann Staudinger, who spent the next decade finding experimental evidence for this hypothesis.
Polymers are of two types: naturally occurring and synthetic or man made.
Natural polymeric materials such as hemp, shellac, amber, wool, silk, and natural rubber have been used for centuries. A variety of other natural polymers exist, such as cellulose, which is the main constituent of wood and paper.
Hemoglycin (previously termed hemolithin) is a space polymer that is the first polymer of amino acids found in meteorites.
The list of synthetic polymers, roughly in order of worldwide demand, includes polyethylene, polypropylene, polystyrene, polyvinyl chloride, synthetic rubber, phenol formaldehyde resin (or Bakelite), neoprene, nylon, polyacrylonitrile, PVB, silicone, and many more. More than 330 million tons of these polymers are made every year (2015).