

Community hub
Recent from talks
Contribute something
Nothing was collected or created yet.
Enolate
View on Wikipedia
In organic chemistry, enolates are organic anions derived from the deprotonation of carbonyl (RR'C=O) compounds. Rarely isolated, they are widely used as reagents in the synthesis of organic compounds.[1][2][3][4]
Bonding and structure
[edit]
Enolate anions are electronically related to allyl anions. The anionic charge is delocalized over the oxygen and the two carbon sites. Thus they have the character of both an alkoxide and a carbanion.[5]
Although they are often drawn as being simple salts, in fact they adopt complicated structures often featuring aggregates.[6]
%3DCMe2(Litmeda)_dimer_(VITLUG).png)
Preparation
[edit]Deprotonation of enolizable ketones, aromatic alcohols, aldehydes, and esters gives enolates.[8][9] With strong bases, the deprotonation is quantitative. Typically enolates are generated from using lithium diisopropylamide (LDA).[10]
Often, as in conventional Claisen condensations, Mannich reactions, and aldol condensations, enolates are generated in low concentrations with alkoxide bases. Under such conditions, they exist in low concentrations, but they still undergo reactions with electrophiles. Many factors affect the behavior of enolates, especially the solvent, additives (e.g. diamines), and the countercation (Li+ vs Na+, etc.). For unsymmetrical ketones, methods exist to control the regiochemistry of the deprotonation.[11]

The deprotonation of carbon acids can proceed with either kinetic or thermodynamic reaction control. For example, in the case of phenylacetone, deprotonation can produce two different enolates. LDA has been shown to deprotonate the methyl group, which is the kinetic course of the deprotonation. To ensure the production of the kinetic product, a slight excess (1.1 equiv) of lithium diisopropylamide is used, and the ketone is added to the base at −78 °C. Because the ketone is quickly and quantitatively converted to the enolate and base is present in excess at all times, the ketone is unable to act as a proton shuttle to catalyze the gradual formation of the thermodynamic product. A weaker base such as an alkoxide, which reversibly deprotonates the substrate, affords the more thermodynamically stable benzylic enolate.
Enolates can be trapped by acylation and silylation, which occur at oxygen. Silyl enol ethers are common reagents in organic synthesis as illustrated by the Mukaiyama aldol reaction:[13]

Role of Lewis acids on enolate formation
[edit]In addition to the use of strong bases, enolates can be generated using a Lewis acid and a weak base ("soft conditions"):

For deprotonation to occur, the stereoelectronic requirement is that the alpha-C-H sigma bond must be able to overlap with the pi* orbital of the carbonyl:

Geometry
[edit]Extensive studies have been performed on the formation of enolates. It is possible to control the geometry of the enolate:[14]

For ketones, most enolization conditions give Z enolates. For esters, most enolization conditions give E enolates. The addition of HMPA is known to reverse the stereoselectivity of deprotonation.

The stereoselective formation of enolates has been rationalized with the Ireland model,[15][16][17][18] although its validity is somewhat questionable. In most cases, it is not known which, if any, intermediates are monomeric or oligomeric in nature; nonetheless, the Ireland model remains a useful tool for understanding enolates.

In the Ireland model, the deprotonation is assumed to proceed by a six-membered or cyclic[19] monomeric transition state. The larger of the two substituents on the electrophile (in the case above, methyl is larger than proton) adopts an equatorial disposition in the favored transition state, leading to a preference for E enolates. The model clearly fails in many cases; for example, if the solvent mixture is changed from THF to 23% HMPA-THF (as seen above), the enolate geometry is reversed, which is inconsistent with this model and its cyclic transition state.
Regiochemistry of enolate formation
[edit]If an unsymmetrical ketone is subjected to base, it has the potential to form two regioisomeric enolates (ignoring enolate geometry). For example:

The trisubstituted enolate is considered the kinetic enolate, while the tetrasubstituted enolate is considered the thermodynamic enolate. The alpha hydrogen deprotonated to form the kinetic enolate is less hindered, and therefore deprotonated more quickly. In general, tetrasubstituted olefins are more stable than trisubstituted olefins due to hyperconjugative stabilization. The ratio of enolate regioisomers is heavily influenced by the choice of base. For the above example, kinetic control may be established with LDA at −78 °C, giving 99:1 selectivity of kinetic: thermodynamic enolate, while thermodynamic control may be established with triphenylmethyllithium at room temperature, giving 10:90 selectivity.
In general, kinetic enolates are favored by cold temperatures, conditions that give relatively ionic metal–oxygen bonding, and rapid deprotonation using a slight excess of a strong, sterically hindered base. The large base only deprotonates the more accessible hydrogen, and the low temperatures and excess base help avoid equilibration to the more stable alternate enolate after initial enolate formation. Thermodynamic enolates are favored by longer equilibration times at higher temperatures, conditions that give relatively covalent metal–oxygen bonding, and use of a slight sub-stoichiometric amount of strong base. By using insufficient base to deprotonate all of the carbonyl molecules, the enolates and carbonyls can exchange protons with each other and equilibrate to their more stable isomer. Using various metals and solvents can provide control over the amount of ionic character in the metal–oxygen bond.
Reactions
[edit]As powerful nucleophiles, enolates react with a variety of electrophiles. The stereoselectivity and regioselectivity is influenced by additives, solvent, counterions, etc. When the electrophiles are alkyl halides, a classic problem arises: O-alkylation vs C-alkylation. Controlling this selectivity has drawn much attention. The negative charge in enolates is concentrated on the oxygen, but that center is also highly solvated, which leads to C-alkylation.[20]
Other important electrophiles are aldehydes/ketones and Michael acceptors.[21]

Sample aldol reaction with lithium enolate

Synthesis of enones using regiospecific enolate formation and masked functionality
[edit]

Regiospecific formation is the controlled enolate formation by the specific deprotonation at one of the α-carbons of the ketone starting molecule. This provides one of the best understood synthetic strategies to introduce chemical complexity in natural product and total syntheses. A prominent example of its use is in the total synthesis of progesterone illustrated in Figure "Regiospecific enolate formation in the total synthesis of progesterone".
When ketones are treated with base, enolates can be formed by deprotonation at either α-carbon. The selectivity is determined by both the steric and electronic effects on the α-carbons as well as the precise base used (see figure ""Masked functionality" for regiospecific enolate formation" for an example of this). Enolate formation will be thermodynamically favoured at the most acidic proton which depends on the electronic stabilization of the resulting anion. However, the selectivity can be reversed by sterically hindering the thermodynamic product and therefore kinetically favouring deprotonation at the other α-carbon centre. Traditional methods for regioselective enolate formation use either electronic activating groups (e.g. aldehydes) or steric blocking groups (e.g. 1,2-ethanedithiol protected ketone).
An enone can also serve as a precursor for regiospecific formation of an enolate, here the enone is a "masked functionality" for the enolate. This process is first described by Gilbert Stork[22] who is best known for his contributions to the study of selective enolate formation methods in organic synthesis. Reacting an enone with lithium metal generates the enolate at the α-carbon of the enone. The enolate product can either be trapped or alkylated. By using "masked functionality", it is possible to produce enolates that are not accessible by traditional methods.
The "masked functionality" approach to regiospecific enolate formation has been widely used in the total synthesis of natural products. For example, in the total synthesis of the steroid hormone progesterone,[23] Stork and co-workers used the "masked functionality" to stereospecifically construct one of the quaternary carbons in the molecule.
Aza enolates
[edit]Aza enolates (also known as imine anions, enamides, metallated Schiff bases, and metalloenamines) are nitrogen analogous to enolates.[24] When imines get treated with strong bases such as LDA, highly nucleophilic aza enolates are generated.
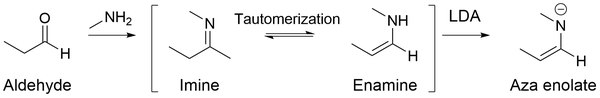
The major benefit of using aza enolates is that they don't undergo self-condensation (i.e. aldol reaction for aldehydes) in a basic or neutral solution, but rather they favor alkylation on the alpha-carbon.[25] This is mainly because imines contain carbon-nitrogen double bonds unlike aldehydes, which contain oxygen-carbon double bonds. Since oxygen is more electronegative than nitrogen, it withdraws more electron density from the carbonyl carbon, inducing a greater partially positive charge on the carbon. Therefore, with more electrophilic carbon, aldehydes allow for better nucleophilic addition to the carbon on the carbon-oxygen double bond.
On the other hand, imine has less electronegative nitrogen which induces a weaker partially positive charge on the carbonyl-carbon. As a result, while imines can still react with organolithiums, they don't react with other nucleophiles (including aza enolates) to undergo nucleophilic additions.[26]
Instead, aza enolates react similarly to enolates, forming SN2 alkylated products.[25] Through nitrogen lone pair conjugation, β-carbon becomes a nucleophilic site, permitting aza enolates to undergo alkylation reactions.[27] Thus, aza enolates can react with numerous electrophiles like epoxides and alkyl halides to form a new carbon-carbon bond on β-carbon.[24]
Two potential reaction mechanisms are shown below:

Since epoxide is a three-membered ring molecule, it has a high degree of ring strain. Although the carbons in the ring system are tetrahedral, preferring 109.5 degrees between each atom, epoxide strains the ring angles into 60 degrees. To counter this effect, the nucleophilic aza enolates easily react with epoxides to reduce their ring strains.

Besides reacting with epoxides, aza enolates can also react with alkyl halides (or allyl halides as depicted above) to form a new carbon-carbon sigma bond. This reaction is one of the key steps in the synthesis of the male aggression pheromone, Oulema melanopus.[29] Aza enolate is generated by LDA reacting with pivaldehyde, which then reacts with an alkyl halide to form an Oulema melanopus intermediate.
Aza enolates can also be formed with Grignard reagents and react with other soft electrophiles, including Michael receptors.[24]
See also
[edit]References
[edit]- ^ Stolz, Daniel; Kazmaier, Uli (2010). "Metal Enolates as Synthons in Organic Chemistry". PATai's Chemistry of Functional Groups. doi:10.1002/9780470682531.pat0423. ISBN 978-0-470-68253-1.
- ^ Hart, David J.; Ha, Deok Chan (1989). "The ester enolate-imine condensation route to .beta.-lactams". Chemical Reviews. 89 (7): 1447–1465. doi:10.1021/cr00097a003.
- ^ Wu, George; Huang, Mingsheng (2006). "Organolithium Reagents in Pharmaceutical Asymmetric Processes". Chemical Reviews. 106 (7): 2596–2616. doi:10.1021/cr040694k. PMID 16836294.
- ^ Curti, Claudio; Battistini, Lucia; Sartori, Andrea; Zanardi, Franca (2020). "New Developments of the Principle of Vinylogy as Applied to π-Extended Enolate-Type Donor Systems". Chemical Reviews. 120 (5): 2448–2612. doi:10.1021/acs.chemrev.9b00481. PMC 7993750. PMID 32040305.
- ^ IUPAC, Compendium of Chemical Terminology, 5th ed. (the "Gold Book") (2025). Online version: (2006–) "Enolates". doi:10.1351/goldbook.E02123
- ^ Reich, Hans J. (2013). "Role of Organolithium Aggregates and Mixed Aggregates in Organolithium Mechanisms". Chemical Reviews. 113 (9): 7130–7178. doi:10.1021/cr400187u. PMID 23941648.
- ^ Nichols, Michael A.; Leposa, Christina M.; Hunter, Allen D.; Zeller, Matthias (2007). "Crystal Structures of Hexameric and Dimeric Complexes of Lithioisobutyrophenone". Journal of Chemical Crystallography. 37 (12): 825–829. doi:10.1007/s10870-007-9255-0. S2CID 97183362.
- ^ Smith, Michael B.; March, Jerry (2007), Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (6th ed.), New York: Wiley-Interscience, ISBN 978-0-471-72091-1
- ^ Manfred Braun (2015). Modern Enolate Chemistry: From Preparation to Applications in Asymmetric Synthesis. Wiley‐VCH. doi:10.1002/9783527671069. ISBN 978-3-527-67106-9.
- ^ Christine Wedler; Hans Schick (1998). "Synthesis of Β-lactones By Aldolization of Ketones with Phenyl Ester Enolates: 3,3-Dimethyl-1-oxaspiro[3.5]nonan-2-one". Org. Synth. 75: 116. doi:10.15227/orgsyn.075.0116.
- ^ Gall, Martin; House, Herbert O. (1972). "The Formation and Alkylation of Specific Enolate Anions from an Unsymmetrical Ketone: 2-Benzyl-2-methylcyclohexanone and 2-Benzyl-6-methylcyclohexanone". Org. Synth. 52: 39. doi:10.15227/orgsyn.052.0039.
- ^ Kong, Jianshe; Meng, Tao; Ting, Pauline; Wong, Jesse (2010). "Preparation of Ethyl 1-Benzyl-4-Fluoropiperidine-4-Carboxylate". Organic Syntheses. 87: 137. doi:10.15227/orgsyn.087.0137.
- ^ Mukaiyama, Teruaki; Kobayashi, Shū (1994). "Tin(II) Enolates in the Aldol, Michael, and Related Reactions". Organic Reactions. pp. 1–103. doi:10.1002/0471264180.or046.01. ISBN 0-471-26418-0.
- ^ Brown, H. C.; Dhar, R. K.; Bakshi, R. K.; Pandiarajan, P. K.; Singaram, B. (1989). "Major effect of the leaving group in dialkylboron chlorides and triflates in controlling the stereospecific conversion of ketones into either E- or Z-enol borinates". Journal of the American Chemical Society. 111 (9): 3441–3442. doi:10.1021/ja00191a058.
- ^ Ireland, R. E.; Willard, A. K. (1975). "The stereoselective generation of ester enolates". Tetrahedron Letters. 16 (46): 3975–3978. doi:10.1016/S0040-4039(00)91213-9.
- ^ Narula, A. S. (1981). "An analysis of the diastereomeric transition state interactions for the kinetic deprotonation of acyclic carbonyl derivatives with lithium diisopropylamide". Tetrahedron Letters. 22 (41): 4119–4122. doi:10.1016/S0040-4039(01)82081-5.
- ^ Ireland, RE; Wipf, P; Armstrong, JD (1991). "Stereochemical control in the ester enolate Claisen rearrangement. 1. Stereoselectivity in silyl ketene acetal formation". Journal of Organic Chemistry. 56 (2): 650–657. doi:10.1021/jo00002a030.
- ^ Xie, L; Isenberger, KM; Held, G; Dahl, LM (October 1997). "Highly Stereoselective Kinetic Enolate Formation: Steric vs Electronic Effects". Journal of Organic Chemistry. 62 (21): 7516–7519. doi:10.1021/jo971260a. PMID 11671880.
- ^ Directed Aldol Synthesis – Formation of E-enolate and Z-enolate
- ^ Smith, Michael B.; March, Jerry (2007), Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (6th ed.), New York: Wiley-Interscience, p. 551, ISBN 978-0-471-72091-1
- ^ Seebach, Dieter (1988). "Structure and Reactivity of Lithium Enolates. From Pinacolone to SelectiveC-Alkylations of Peptides. Difficulties and Opportunities Afforded by Complex Structures". Angewandte Chemie International Edition in English. 27 (12): 1624–1654. doi:10.1002/anie.198816241.
- ^ Stork, G.; Singh, J., J. Am. Chem. Soc. 1974, 96, 6181.
- ^ Stork, G.; McMurry, J. E., J. Am. Chem. Soc. 1967, 89, 5464.
- ^ a b c Aslam, O. (28 September 2012). Development of catalytic aza enolate reactions (Doctoral). UCL (University College London).
- ^ a b Clayden, Jonathan (2012). Organic chemistry (2nd ed.). Oxford: Oxford University Press. pp. 465, 593–594. ISBN 978-0-19-927029-3.
- ^ Cranwell, Philippa. "Enamines/aza-enolates – Mechanism Mordor". sites.google.com. Archived from the original on 2021-09-03. Retrieved 2020-11-28.
- ^ Carey, Francis A. (2007). Advanced organic chemistry. Part B, Reactions and synthesis (5th ed.). New York, NY: Springer. pp. 46–47. ISBN 978-0-387-68350-8.
- ^ Hudrlik, Paul F.; Wan, Chung-Nan (October 1975). "Reactions of oxetane with imine salts derived from cyclohexanone". The Journal of Organic Chemistry. 40 (20): 2963–2965. doi:10.1021/jo00908a027.
- ^ a b Chevalley, Alice; Férézou, Jean-Pierre (2012). "One-pot formation of aza-enolates from secondary amines and condensation to esters and alkyl bromides". Tetrahedron. 68 (29): 5882–5889. doi:10.1016/j.tet.2012.04.105.