Recent from talks
Persistent truncus arteriosus
Knowledge base stats:
Talk channels stats:
Members stats:
Persistent truncus arteriosus
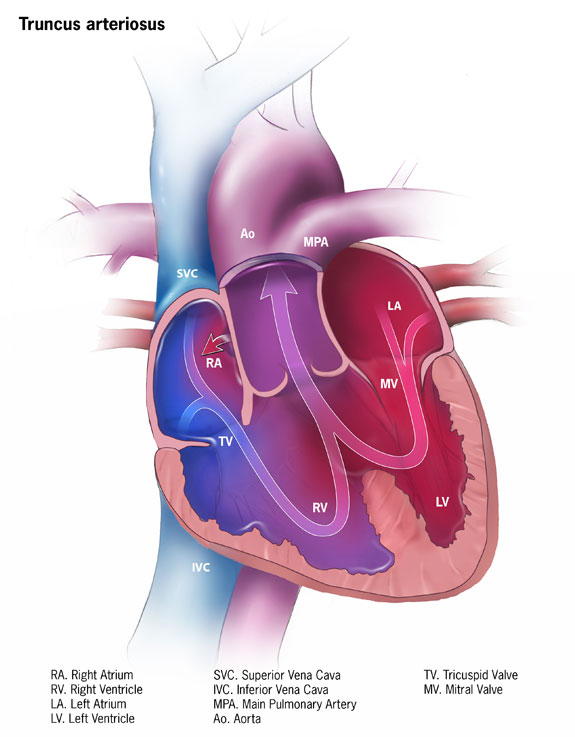
Persistent truncus arteriosus (PTA), often referred to simply as truncus arteriosus, is a rare form of congenital heart disease that presents at birth. In this condition, the embryological structure known as the truncus arteriosus fails to properly divide into the pulmonary trunk and aorta. This results in one arterial trunk arising from the heart and providing mixed blood to the coronary arteries, pulmonary arteries, and systemic circulation. For the International Classification of Diseases (ICD-11), the International Paediatric and Congenital Cardiac Code (IPCCC) was developed to standardize the nomenclature of congenital heart disease. Under this system, English is now the official language, and persistent truncus arteriosus should properly be termed common arterial trunk.
Most of the time, this defect occurs spontaneously. Genetic disorders and teratogens (viruses, metabolic imbalance, and industrial or pharmacological agents) have been associated as possible causes. Up to 50% (varies in studies) of cases are associated with chromosome 22q11 deletions (DiGeorge Syndrome). The neural crest, specifically a population known as the cardiac neural crest, directly contributes to the aorticopulmonary septum.
Microablation of the cardiac neural crest in developing chick embryos and genetic anomalies affecting this population of cells in rodents results in persistent truncus arteriosus.
Numerous perturbations affecting the cardiac neural crest have been associated with persistent truncus arteriosus, some of which include growth factors (fibroblast growth factor 8 and bone morphogenetic protein), transcription factors (T-box, Pax, Nkx2-5, GATA-6, and Forkhead), and gap junction proteins (Connexin). The cardiac neural crest also contributes the smooth muscle of the great arteries.[citation needed]
Anatomical changes associated with this disorder includes:[citation needed]
The diagnosis is based on:[citation needed]
A well-known classification is the fourfold system developed by Collett and Edwards in 1949. Collett/Edwards Types I, II, and III are distinguished by the branching pattern of the pulmonary arteries:
Another well-known classification was defined by Stella and Richard Van Praagh in 1965. In this classification scheme, the preceding letter ("A" or "B") refers to the presence or absence, respectively, of a ventricular septal defect. Type B common arterial trunk is extremely rare; so below, only Type A is considered:[citation needed]
Hub AI
Persistent truncus arteriosus AI simulator
(@Persistent truncus arteriosus_simulator)
Persistent truncus arteriosus
Persistent truncus arteriosus (PTA), often referred to simply as truncus arteriosus, is a rare form of congenital heart disease that presents at birth. In this condition, the embryological structure known as the truncus arteriosus fails to properly divide into the pulmonary trunk and aorta. This results in one arterial trunk arising from the heart and providing mixed blood to the coronary arteries, pulmonary arteries, and systemic circulation. For the International Classification of Diseases (ICD-11), the International Paediatric and Congenital Cardiac Code (IPCCC) was developed to standardize the nomenclature of congenital heart disease. Under this system, English is now the official language, and persistent truncus arteriosus should properly be termed common arterial trunk.
Most of the time, this defect occurs spontaneously. Genetic disorders and teratogens (viruses, metabolic imbalance, and industrial or pharmacological agents) have been associated as possible causes. Up to 50% (varies in studies) of cases are associated with chromosome 22q11 deletions (DiGeorge Syndrome). The neural crest, specifically a population known as the cardiac neural crest, directly contributes to the aorticopulmonary septum.
Microablation of the cardiac neural crest in developing chick embryos and genetic anomalies affecting this population of cells in rodents results in persistent truncus arteriosus.
Numerous perturbations affecting the cardiac neural crest have been associated with persistent truncus arteriosus, some of which include growth factors (fibroblast growth factor 8 and bone morphogenetic protein), transcription factors (T-box, Pax, Nkx2-5, GATA-6, and Forkhead), and gap junction proteins (Connexin). The cardiac neural crest also contributes the smooth muscle of the great arteries.[citation needed]
Anatomical changes associated with this disorder includes:[citation needed]
The diagnosis is based on:[citation needed]
A well-known classification is the fourfold system developed by Collett and Edwards in 1949. Collett/Edwards Types I, II, and III are distinguished by the branching pattern of the pulmonary arteries:
Another well-known classification was defined by Stella and Richard Van Praagh in 1965. In this classification scheme, the preceding letter ("A" or "B") refers to the presence or absence, respectively, of a ventricular septal defect. Type B common arterial trunk is extremely rare; so below, only Type A is considered:[citation needed]
Recent media