

Community hub
Recent from talks
Contribute something
Nothing was collected or created yet.
Astrocytoma
View on Wikipedia| Astrocytoma | |
|---|---|
 | |
| Two PET images—the upper of which shows a normal brain and the lower shows astrocytoma | |
| Specialty | Neuro-oncology, neurosurgery |
Astrocytoma is a type of brain tumor. Astrocytomas (also astrocytomata) originate from a specific kind of star-shaped glial cell in the cerebrum called an astrocyte. This type of tumor does not usually spread outside the brain and spinal cord, and it does not usually affect other organs. After glioblastomas, astrocytomas are the second most common glioma and can occur in most parts of the brain and occasionally in the spinal cord.[1]
Within the astrocytomas, two broad classes are recognized in literature, those with:
- Narrow zones of infiltration (mostly noninvasive tumors; e.g., pilocytic astrocytoma, subependymal giant cell astrocytoma, pleomorphic xanthoastrocytoma), that often are clearly outlined on diagnostic images
- Diffuse zones of infiltration (e.g., high-grade astrocytoma), that share various features, including the ability to arise at any location in the central nervous system, but with a preference for the cerebral hemispheres; they occur usually in adults, and have an intrinsic tendency to progress to more advanced grades.[2]
People can develop astrocytomas at any age. The low-grade type is more often found in children or young adults, while the high-grade type is more prevalent in adults. Astrocytomas in the base of the brain are more common in young people and account for roughly 75% of neuroepithelial tumors.[3]
Pathophysiology
[edit]Astrocytoma causes regional effects by compression, invasion, and destruction of brain parenchyma, arterial and venous hypoxia, competition for nutrients, release of metabolic end products (e.g., free radicals, altered electrolytes, neurotransmitters), and release and recruitment of cellular mediators (e.g., cytokines) that disrupt normal parenchymal function.[2] Secondary clinical sequelae may be caused by elevated intracranial pressure attributable to direct mass effect, increased blood volume, or increased cerebrospinal fluid volume.[2]
Genetic and Molecular alterations
[edit]Homozygous deletion of CDKN2A/B is the main feature of high grade astrocytoma. In addition, a genome-wide pattern of DNA copy-number alterations (CNAs) has been uncovered, which is correlated with a patient's survival and response to treatment. This pattern identifies among lower-grade astrocytoma patients a subtype, where the CNA genotype is correlated with an approximately one-year survival phenotype.[4][5]
Diagnosis
[edit]An X-ray computed tomography (CT) or magnetic resonance imaging (MRI) scan is necessary to characterize the extent of these tumors (size, location, consistency). CT will usually show distortion of third and lateral ventricles with displacement of anterior and middle cerebral arteries. Histologic analysis is necessary for grading diagnosis.[citation needed]
In the first stage of diagnosis the doctor will take a history of symptoms and perform a basic neurological exam, including an eye exam and tests of vision, balance, coordination, and mental status. The doctor will then require a CT scan and MRI of the patient's brain. During a CT scan, X-rays of the patient's brain are taken from many different directions. These are then combined by a computer, producing a cross-sectional image of the brain. For an MRI, the patient relaxes in a tunnel-like instrument while the brain is subjected to changes of magnetic field. An image is produced based on the behavior of the brain's water molecules in response to the magnetic fields. A special dye may be injected into a vein before these scans to provide contrast and make tumors easier to identify.[citation needed]
If a tumor is found, a neurosurgeon must perform a biopsy on it. This simply involves the removal of a small amount of tumor tissue, which is then sent to a neuropathologist for examination and grading. The biopsy may take place before surgical removal of the tumor or the sample may be taken during surgery. Grading of the tumor sample is a method of classification that helps the doctor to determine the severity of the astrocytoma and to decide on the best treatment options. The neuropathologist grades the tumor by looking for atypical cells, the growth of new blood vessels, and for indicators of cell division called mitotic figures.[citation needed]
-
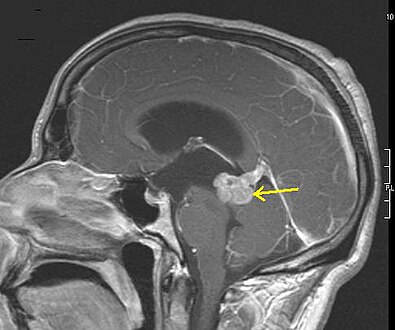 Low grade astrocytoma of the midbrain (lamina tecti), sagittal T1-weighted magnetic resonance imaging after contrast medium administration: The tumor is marked with an arrow. The CSF spaces in front of the tumor are expanded due to compression-induced hydrocephalus internus.
Low grade astrocytoma of the midbrain (lamina tecti), sagittal T1-weighted magnetic resonance imaging after contrast medium administration: The tumor is marked with an arrow. The CSF spaces in front of the tumor are expanded due to compression-induced hydrocephalus internus. -
 A pathological specimen of a gemistocytic astrocytoma
A pathological specimen of a gemistocytic astrocytoma -
 MRI scans of an astrocytoma patient, showing tumor progression over the course of seven years
MRI scans of an astrocytoma patient, showing tumor progression over the course of seven years



Grading
[edit]Of numerous grading systems in use for the classification of tumor of the central nervous system, the World Health Organization (WHO) grading system is commonly used for astrocytoma. Established in 1993 in an effort to eliminate confusion regarding diagnoses, the WHO system established a four-tiered histologic grading guideline for astrocytomas that assigns a grade from 1 to 4, with 1 being the least aggressive and 4 being the most aggressive.[citation needed]
The WHO grading scheme is based on the appearance of certain characteristics: atypia, mitosis, endothelial proliferation, and necrosis. These features reflect the malignant potential of the tumor in terms of invasion and growth rate. Various types of astrocytomas are given these WHO grades:
| WHO grade | Astrocytomas | Description |
|---|---|---|
| I | Consist of slow-growing astrocytomas, benign, and associated with long-term survival. Individuals with very slow-growing tumors where complete surgical removal by stereotactic surgery is possible may experience total remission.[6] Even if the surgeon is not able to remove the entire tumor, it may remain inactive or be successfully treated with radiation. | |
| II |
|
Consist of relatively slow-growing astrocytomas, usually considered benign that sometimes evolve into more malignant or as higher grade tumors. They are prevalent in younger people who often present with seizures. Median survival varies with the cell type of the tumor. Grade 2 astrocytomas are defined as being invasive gliomas, meaning that the tumor cells penetrate into the surrounding normal brain, making a surgical cure more difficult. People with oligodendrogliomas (which might share common cells of origin[7]) have better prognoses than those with mixed oligoastrocytomas, who in turn have better prognoses than patients with (pure) low-grade astrocytomas. Other factors which influence survival include age (younger the better) and performance status (ability to perform tasks of daily living). Due to the infiltrative nature of these tumors, recurrences are relatively common. Depending on the patient, radiation or chemotherapy after surgery is an option. Individuals with grade 2 astrocytoma have a 5-year survival rate of about 34% without treatment and about 70% with radiation therapy.[6] The median survival time is 4 years.[6] |
| III | Consist of anaplastic astrocytomas. It is often related to seizures, neurologic deficits, headaches, or changes in mental status. The standard initial treatment is to remove as much of the tumor as possible without worsening neurologic deficits. Radiation therapy has been shown to prolong survival and is a standard component of treatment. Individuals with grade 3 astrocytoma have a median survival time of 18 months without treatment (radiation and chemotherapy).[6] There is no proven benefit to adjuvant chemotherapy or supplementing other treatments for this kind of tumor. Although temozolomide is effective for treating recurrent anaplastic astrocytoma, its role as an adjuvant to radiation therapy has not been fully tested. | |
| IV |
|
Consists of grade 4 astrocytoma (as of WHO 2021) that form following high-grade transformation of low-grade astrocytoma. These are more common in younger patients (mean age 45 versus 62 years).[7] Surgical removal remains the mainstay of treatment, provided that unacceptable neurologic injury can be avoided. The extremely infiltrative nature of this tumor makes complete surgical removal impossible. Although radiotherapy rarely cures glioblastoma, studies show that it doubles the median survival of patients, compared to supportive care alone.[8] The prognosis is worst for these grade 4 gliomas. Few patients survive beyond 3 years. Individuals with grade 4 astrocytoma have a median survival time of 17[6] weeks without treatment, 30[6] weeks with radiation, and 37[6] weeks with surgical removal of most of the tumor followed by radiation therapy. Long-term survival (at least five years) falls well under 3%.[9][10] |

According to the WHO data, the lowest grade astrocytomas (grade I) make up only 2% of recorded astrocytomas, grade II 8%, and the higher grade anaplastic astrocytomas (grade III) 20%. The highest graded astrocytoma (grade IV GBM) is the most common primary nervous system cancer and second most frequent brain tumor after brain metastasis. Despite the low incidence of astrocytomas compared to other human cancers, mortality is significant, as the higher grades (III & IV) present high mortality rates (mainly due to late detection of the neoplasm).[citation needed]
Prevention
[edit]There are no precise guidelines because the exact cause of astrocytoma is not known.[citation needed]
Treatment
[edit]
For low-grade astrocytomas, removal of the tumor generally allows functional survival for many years. In some reports, the 5-year survival has been over 90% with well-resected tumors. Indeed, broad intervention of low-grade conditions is a contested matter. In particular, pilocytic astrocytomas are commonly indolent bodies that may permit normal neurologic function. However, left unattended, these tumors may eventually undergo neoplastic transformation. To date, complete resection of high-grade astrocytomas is impossible because of the diffuse infiltration of tumor cells into normal parenchyma. Thus, high-grade astrocytomas inevitably recur after initial surgery or therapy and are usually treated similarly to the initial tumor. Despite decades of therapeutic research, curative intervention is still nonexistent for high-grade astrocytomas; patient care ultimately focuses on palliative management.[3]
Society and culture
[edit]Notable cases
[edit]In March 1990, United States Republican Party political strategist Lee Atwater was diagnosed with astrocytoma after a tumor was found in his right parietal lobe. After undergoing radiation therapy (including the then-new implant radiation treatment), Atwater died the following year at the age of 40.[12]
Long-time U.S. Senator Ted Kennedy (D-MA) died of malignant glioma.[13]
University of Texas sniper Charles Whitman, who killed multiple people during a mass murder event in 1966, was diagnosed with astrocytoma post-mortem. The Connally Commission investigating the shooting disagreed, and identified it as a glioblastoma, and concluded the tumor "conceivably could have contributed to his inability to control his emotions and actions".[14]
Major League pitcher Dan Quisenberry was diagnosed with grade IV astrocytoma in January 1998. He died at the age of 45 in 1998 in Leawood, Kansas.[15]
Richard Burns, winner of the 2001 World Rally Championship, was diagnosed with astrocytoma in 2003. Four years to the day after winning the World Rally Championship, on 25 November 2005, Burns died in Westminster, London,[16] aged 34, after having been in a coma for some days as a result of his brain tumour.[17]
Professional wrestler Matt Cappotelli was diagnosed with a grade 2/3 astrocytoma in December 2005, scuttling plans to promote Cappottelli to the main WWE roster. Cappotelli, who won a contract with WWE through the third season of their reality program Tough Enough, was the Ohio Valley Wrestling Heavyweight Champion at the time of his diagnosis and vacated the title in February 2006[18] after confirming the tumor was cancerous. Cappotelli underwent successful surgery and chemotherapy,[19][20][21] but was unable to return to active wrestling work. He did return to OVW as a trainer in 2013.[22][23] He died on June 29, 2018.[24]
Kelley Mack was an American actress.[25] She played Addy in season 9 of the series The Walking Dead (2018–2019). She also had roles in the films Profile (2018) and Broadcast Signal Intrusion (2021). In January 2025, Mack announced she had been diagnosed with astrocytoma.[26][27] By April 2025, she had completed proton radiation treatment.[25] Mack died in Cincinnati on August 2, 2025, at the age of 33.[28][29][30]
References
[edit]- ^ "What Is Astrocytoma?". WebMD. Retrieved 1 July 2021.
- ^ a b c Astrocytoma at eMedicine
- ^ a b "Astrocytoma". The Lecturio Medical Concept Library. Retrieved 1 July 2021.
- ^ C. H. Lee*; B. O. Alpert*; P. Sankaranarayanan; O. Alter (January 2012). "GSVD Comparison of Patient-Matched Normal and Tumor aCGH Profiles Reveals Global Copy-Number Alterations Predicting Glioblastoma Multiforme Survival". PLOS ONE. 7 (1) e30098. Bibcode:2012PLoSO...730098L. doi:10.1371/journal.pone.0030098. PMC 3264559. PMID 22291905. Highlight.
- ^ S. P. Ponnapalli, et int.; O. Alter (May 2020). "Retrospective Clinical Trial Experimentally Validates Glioblastoma Genome-Wide Pattern of DNA Copy-Number Alterations Predictor of Survival". APL Bioengineering. 4 (2): 026106. doi:10.1063/1.5142559. PMC 7229984. PMID 32478280. Press Release.
- ^ a b c d e f g mdguidelines.com Astrocytoma Archived 2017-11-30 at the Wayback Machine Retrieved on Mars 26, 2010
- ^ a b Ohgaki, Hiroko; Kleihues, Paul (2009). "Genetic alterations and signaling pathways in the evolution of gliomas". Cancer Science. 100 (12): 2235–41. doi:10.1111/j.1349-7006.2009.01308.x. PMC 11159448. PMID 19737147. S2CID 16742915.
- ^ "Glioma - Symptoms and causes". Mayo Clinic.
- ^ Buckner, Jan C.; Brown, Paul D.; O'Neill, Brian P.; Meyer, Fredric B.; Wetmore, Cynthia J.; Uhm, Joon H. (2007). "Central Nervous System Tumors". Mayo Clinic Proceedings. 82 (10): 1271–86. doi:10.4065/82.10.1271. PMID 17908533.
- ^ "CBTRUS - CBTRUS". CBTRUS.
- ^ a b Weller M, van den Bent M, Preusser M, Le Rhun E, Tonn JC, Minniti G; et al. (2021). "EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood". Nat Rev Clin Oncol. 18 (3): 170–186. doi:10.1038/s41571-020-00447-z. PMC 7904519. PMID 33293629.
{{cite journal}}: CS1 maint: multiple names: authors list (link)
"This article is licensed under a Creative Commons Attribution 4.0 International License" - ^ Brady, John (December 1, 1996). "I'm Still Lee Atwater", The Washington Post, retrieved 2010-04-11.
- ^ "Kennedy fought aggressive cancer". CNN. August 26, 2009. Retrieved 2010-02-27.
- ^ Waring, Thomas R., ed. "Jury Blames Tumor For Killings: Doctor Says Whitman Unaffected"" The News and Courier [Charleston] 05 Aug. 1966: 9B. Print.
- ^ Henderson, Heather (1999). "Dan Quisenberry - In His Own Words" The 1999 Big Bad Baseball Annual. Retrieved June 24, 2013.
- ^ "Deaths England and Wales 1984–2006". Archived from the original on 2009-08-31. Retrieved 2015-11-25.
- ^ "Former world champion Burns dies". bbc.co.uk. 26 November 2005. Retrieved 25 February 2008.
- ^ Brady, Hicks. "2006: The year in wrestling". PWI Presents: 2007 Wrestling Almanac and book of facts. Kappa Publications. p. 17. 2007 Edition.
- ^ Dee, Louie (2007-04-06). "Matt Cappotelli to undergo brain surgery". WWE.
- ^ "Cappotelli undergoes surgery". World Wrestling Entertainment. 2007-05-01. Retrieved 2015-08-26.
- ^ "Cappotelli recovering well". World Wrestling Entertainment. 2007-05-09.
- ^ "Helping wrestlers get a grip". The Courier-Journal. Louisville, KY. 2013-11-12. Retrieved 2015-08-26.
- ^ "Matt Cappotelli Beginner's Program". Ohio Valley Wrestling. Archived from the original on 2015-08-20. Retrieved 2015-08-26.
- ^ Xu, Linda (June 29, 2018). "Matt Cappotelli, Former WWE Wrestler and 'Tough Enough' Winner, Dies at 38". TheWrap. Retrieved June 29, 2018.
- ^ a b Robinson, KiMi. "Kelley Mack, 'The Walking Dead' and 'Chicago Med' actress, dies at 33". USA Today. Gannett. ISSN 0734-7456. OCLC 8799626. Retrieved August 6, 2025.
- ^ Cite error: The named reference
Moseswas invoked but never defined (see the help page). - ^ Rudoy, Matthew (August 6, 2025). "The Walking Dead Star Kelley Mack Passes Away At The Age Of 33". Screen Rant. Valnet. Retrieved August 6, 2025.
- ^ Saunders, Angel (August 5, 2025). "'The Walking Dead' Actress Kelley Mack Dies at 33". People. People Inc. ISSN 0093-7673. OCLC 794712888. Retrieved August 5, 2025.
- ^ Mendoza, Leia (August 5, 2025). "Kelley Mack, '9-1-1' and 'The Walking Dead' Actress, Dies At 33". Variety. Penske Media Corporation. ISSN 0042-2738. OCLC 60626328. Retrieved August 6, 2025.
- ^ Memetz, Dave (August 5, 2025). "The Walking Dead's Kelley Mack dead at 33". TVLine. TVLine Media, LLC. Retrieved August 6, 2025.
Bibliography
- "Astrocytomas." KidsHealth - the Web's most visited site about children's health. Web. 01 May 2024. KidsHealth.org - Nemours KidsHealth.
- "Astrocytoma - Diagnosis and Treatment Options at Mayo Clinic." Mayo Clinic: Medical Treatment and Research Centers. Web. 07 Dec. 2009. Glioma - Symptoms and causes.
- "Glioblastoma Multiforme Treatment at Mayo Clinic." Mayo Clinic: Medical Treatment and Research Centers. Web. 07 Dec. 2009. Glioma - Symptoms and causes.
- "The new WHO Classification of Tumors affecting the Central Nervous System" by Stephen B. Tatter, M.D., Ph.D.; MGH Archived 2012-01-30 at the Wayback Machine
External links
[edit]- Cancer.Net: Astrocytoma, Childhood
- Imaging Astrocytoma MR, CT, Pathology