
Community hub
Recent from talks
Contribute something
Nothing was collected or created yet.
Aggressive fibromatosis
View on WikipediaYou can help expand this article with text translated from the corresponding article in Polish. (August 2023) Click [show] for important translation instructions.
|
| Aggressive fibromatosis | |
|---|---|
| Other names | Desmoid tumor, deep fibromatosis, desmoid fibromatosis |
 | |
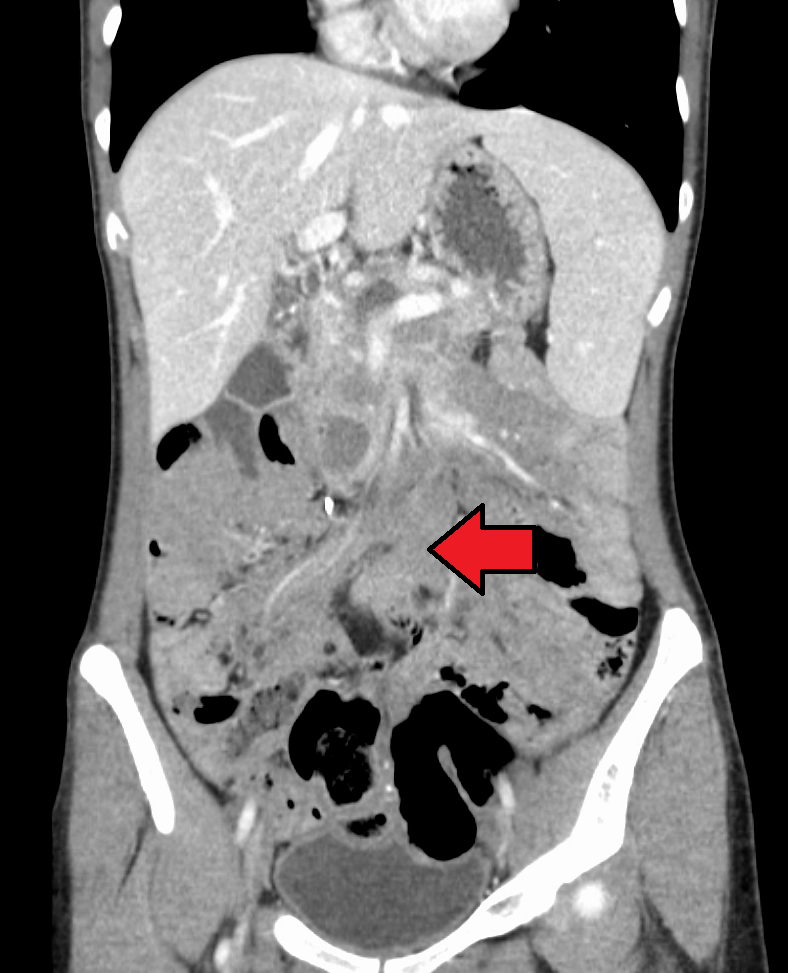
| Desmoid tumor as seen on CT scan | |
| Specialty | Oncology, surgery, radiology |
| Complications | Pain, loss of function, restricted movement |
| Usual onset | 30–40 years[1] |
| Risk factors | CTNNB1 and APC gene mutations, familial adenomatous polyposis, estrogen levels, pregnancy, physical trauma or surgery |
| Diagnostic method | Biopsy |
| Differential diagnosis | Broad, including fibroblastic sarcomas, superficial fibromatosis, nodular fasciitis, gastrointestinal stromal tumor, and scar tissue |
| Treatment | Watchful waiting; surgery; radiation therapy‚ chemotherapy; antiestrogen medication; NSAIDs; ablation with cold, heat, or ultrasound |
| Incidence | 5–6 per million per year[2] |
Aggressive fibromatosis or desmoid tumor is a rare condition. Desmoid tumors are a type of fibromatosis and related to sarcoma, though without the ability to spread throughout the body (metastasize). The tumors arise from cells called fibroblasts, which are found throughout the body. Fibroblasts provide protection to the vital organs and structural support to other tissues, and play a critical role in wound healing. Desmoid tumors tend to occur in women in their thirties, but can occur in anyone at any age. They can be either relatively slow-growing or malignant. However, aggressive fibromatosis is locally aggressive and invasive, with spindle-like growths. The tumors can lead to pain, life-threatening problems, or, rarely, death when they invade other soft tissue or compress vital organs such as intestines, kidneys, lungs, blood vessels, or nerves. Most cases are sporadic, but some are associated with familial adenomatous polyposis (FAP). Approximately 10% of individuals with Gardner's syndrome, a type of FAP with extracolonic features, have desmoid tumors.[3]
In 2020, the World Health Organization reclassified desmoid tumors (termed desmoid-type fibromatosis) as a specific type of tumor in the category of intermediate (locally aggressive) fibroblastic and myofibroblastic tumors.[4]
Histologically they resemble very low-grade fibrosarcomas,[5] but they are very locally aggressive and tend to recur even after complete resection. The condition is "characterized by a variable and often unpredictable clinical course."[2] There is a tendency for recurrence in the setting of prior surgery; in one study, two-thirds of patients with desmoid tumors had a history of prior abdominal surgery.[6] The condition can be chronic and may be debilitating.[7]
Causes
[edit]Wnt signaling pathway alterations are the likely cause of desmoid tumor formation.[8] Mutations have been discovered in both the beta-catenin encoding CTNNB1 gene and the tumor-suppressing APC gene, which affect the Wnt pathway. A 2015 study on desmoid tumors lacking these mutations found that almost all, 95%, "may have mutations that affect the Wnt/β-catenin pathway, suggesting a near universal relationship between desmoid tumors and Wnt signaling."[8]
The majority of cases are sporadic, most of which – 85% – involve a CTNNB1 mutation.[9] Of these, "the three distinct mutations identified are 41A, 45F, and 45. Mutation 45F is associated with a high risk of recurrence."[1] APC mutations affect FAP patients and make up a smaller percentage, 10–15%, of sporadic cases.[9]
The disease has a tendency to occur during and after pregnancy and in exposure to higher estrogen levels, suggesting a hormonal link.[10] One study noted the formation of desmoid tumors in guinea pigs after prolonged estrogen exposure.[11] Other factors include trauma and surgery.[8]
Risk factors for desmoid disease amongst FAP patients include female sex, a 3' APC mutation, a positive family history, and a history of previous abdominal surgery.[12]
Diagnosis
[edit]A biopsy is always indicated as the definitive method to determine the nature of the tumor.[1] Diagnosis may be difficult in part due to the use of core needle biopsy over open biopsy.[13]
Similarities among bland spindle-cell lesions lead to a large number of possibilities in diagnosis, including fibroblastic sarcomas, Gardner fibroma, scar tissue or keloids, superficial fibromatosis, nodular fasciitis, myofibroma, collagenous fibroma, gastrointestinal stromal tumor, solitary fibrous tumor, phyllodes tumor, and other conditions. Such conditions may therefore also be incorrectly diagnosed as desmoid tumors (29% of cases in one review).[14][10][15][16] Some 30–40% of desmoid tumors may be misdiagnosed.[17]
Classification
[edit]

Desmoid tumors can occur almost anywhere in the body.[18] They are classified as extra-abdominal, abdominal wall, or intra-abdominal; the last is more common in patients with FAP.[19] Most cases occur in the mesentery, abdominal wall, and extremities.[20] One study has shown extra-abdominal tumors making up 43% of cases, abdominal tumors 49%, and mesenteric 8%, though statistics vary.[11] Pregnancy-related tumors typically arise in the abdominal wall.[21] Tumors located intra-abdominally or in the head and neck have the highest risk of mortality due to the proximity to vital structures.[22]
One analysis has shown a median tumor size of 7.5 cm (3.0 in).[18] Though metastasis cannot occur, the tumors may in some cases be multifocal, with several located in the same body part.[23]
A 3' APC mutation is the most significant risk factor for intra-abdominal desmoid development amongst FAP patients.[24] FAP patients presenting with an abdominal wall desmoid pre-operatively are at an increased risk of developing an intra-abdominal desmoid post-operatively.[25]
Desmoid tumors of the breast are rare, constituting 4% of extra-abdominal cases and 0.2–0.3% of breast tumors.[20][16] Although benign, they can mimic breast cancer on physical examination, mammography and breast ultrasound and can also be locally invasive. Even though they occur sporadically, they can also be seen as a part of Gardner's syndrome. Some cases – up to 44% – occur in patients who have previously had breast surgery.[26] A high index of suspicion and a thorough triple examination protocol is necessary to detect rare lesions like a desmoid tumor which can masquerade as breast carcinoma. Desmoid tumor of the breast may present a difficulty in the diagnosis especially where imaging studies are not conclusive and suggest a more ominous diagnosis.[27] They may arise in the chest wall or the breast itself.[11][16]
Desmoid tumors may occur in the head and neck, more commonly among children, and tend to be more aggressive than in other extra-abdominal locations. These tumors constitute up to 23% of extra-abdominal cases.[11] Treatment is typically more aggressive due to the increased dangers of a tumor in the area.[23][28]
Staging
[edit]There is no standard staging system; desmoid tumors do not fall under cancer staging systems as they do not metastasize.[26]
Treatment
[edit]Nirogacestat, a selective gamma secretase inhibitor, was approved for medical use in the United States in November 2023.[29] It is the first medication approved by the US Food and Drug Administration (FDA) for the treatment of desmoid tumors.[29][30]
A Phase 2/3 trial on AL102, another selective gamma secretase inhibitor, is also ongoing as of 2023[update], having begun in 2021.[31] The drug was granted orphan drug status in 2023.[32]
Wnt pathway inhibitors are also being developed and studied as of 2024[update]. These include E7386, tegavivint and ipafricept.[33] Additionally, the tumor microenvironment in desmoid tumors is being investigated to find new targets for treatment.[34]
Surgery was the standard treatment for desmoid tumors up to the early 2000s.[2][35] Due to the condition's unpredictability, more conservative management such as watchful waiting has since become common due to the potential impacts of surgical interventions. As of the 2010s, there is a "clear consensus"[2] from medical groups, including the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group and the European Society for Medical Oncology: immediate surgical resection is no longer the first-line treatment, particularly in asymptomatic patients.[8][20][2] Complete removal is not always possible due to the tumors' infiltrative nature and tendril-like growth.[10]
In more advanced, recurring, or rapidly progressing cases, treatment may consist of complete surgical removal, radiation therapy, antiestrogens (e.g. tamoxifen), nonsteroidal anti-inflammatory drugs (NSAIDs), chemotherapy (e.g. methotrexate and vinblastine or vinorelbine, doxorubicin), or ablation (cold, heat, ultrasound). Treatment with oral tyrosine kinase inhibitor drugs (e.g. imatinib, sorafenib, pazopanib, sunitinib) shows promising success rates.[36][23][37][33][38] Radiation therapy after surgery may improve outcomes.[10][38] Despite the condition's hormonal link, anti-hormonal therapies only appear to work in a small subset of patients.[10]
Intestinal transplant is a treatment option for those patients with complicated desmoid tumor, such as those involving the mesenteric root, or those with intestinal failure resulting from the tumor or prior interventions.[39]
MRI or CT imaging scans are commonly used for monitoring.[40][1]
In contrast with cancer, management of desmoid tumors considers additional outcomes beyond progression-free survival and overall survival, as desmoid tumor patients' "survival is longer and ... age of onset is generally younger compared with cancer patient populations".[41]
Outcomes
[edit]Disease course
[edit]The condition is "characterized by a variable and often unpredictable clinical course",[2] often considered chronic,[8] and with the potential to be debilitating.[7] Death, however, is uncommon.[22][41] Tumors may grow, regress, or remain stable:[17]
- Resolution without treatment (10–28%)
- Progression and resolution (30%)
- Stable (50%)
- Rapid progression (10%)
Management of these lesions is complex, the main problem being the high rates of recurrence particularly in FAP-associated disease. Recurrence rates in general vary from 19 to 77 percent.[11] Conversely, for intra-abdominal fibromatosis without evidence of FAP, although extensive surgery may still be required for local symptoms, the risk of recurrence appears to be lower.[42]
Impacts
[edit]One review summarizes the disease's impact on patients stating, "the burden of [desmoid tumors] is disproportionately borne by women of childbearing and working age, and because it is associated with low mortality and a relatively young patient population, it typically continues for decades."[41]
Symptoms vary significantly as they are dependent on the tumor's location and effects on the surrounding structures.[41] Though desmoid tumors do not metastasize, their invasiveness may lead to pain and loss of function or restricted movement. Chronic pain is an issue for as many as 63% of patients and may be debilitating and lead to reliance on pain medication.[17][41] Pressure on vital organs or deformity may occur.[17][10] Rarely, amputation may be necessary due to injury caused by the tumor or its treatments.[41]
Tumors may be misdiagnosed (30–40%)[17] due to their rarity and a lack of knowledge; patients may initially be given inappropriate treatment or poor prognoses due to misdiagnosis with conditions such as malignant sarcoma.[43][44] Patients may need to visit multiple healthcare providers to receive a diagnosis, causing delay in care. Patients may experience issues including anxiety, fatigue, or trouble sleeping; despite the increased survival rate, their level of emotional distress has been compared to that of cancer patients, including "patients with sarcoma, also a malignant connective tissue disorder".[17][44][41] A lack of knowledge by healthcare providers and of information available to patients and others have also been cited as issues.[43]
The economic burden of treatment may be significant, with surgery costs estimated at $50,000 in 2022 US dollars.[35]
Specific instruments to determine health-related quality of life impacts for desmoid patients, the Gounder/Desmoid Tumor Research Foundation (DTRF) Desmoid Symptom/Impact Scale (GODDESS) and the Desmoid-type fibromatosis Quality of Life Questionnaire (DTF-QOL) have been developed and validated.[17]
Epidemiology
[edit]The incidence of desmoid tumors is 5–6 per million per year;[2] they constitute 0.03% of tumors and less than 3% of soft-tissue tumors. The primary age range is 15–60, with a peak between 30 and 40 years old; it is 2–3 times more common in females than males.[1][45][41] A 2012 retrospective multi-institutional analysis of 211 patients found a median age of 36 and a 68% female prevalence.[18] Children do not have the same sex disparity and are most commonly affected around 15 or 16 years old.[22]
History and etymology
[edit]
The condition was first described in 1832 by John MacFarlane. Desmoid, used by Johannes Peter Müller in 1838, comes from the Greek desmos 'band or tendon-like', describing the tumors' consistency.[45][46] The term found broad acceptance in the 1880s.[47] Over the next several decades, Georg Ledderhose and C. Pfeiffer compiled and reported a number of cases, reaching 400 by the early 1900s.[47] In 1923, Ralph W. Nichols first described the correlation between familial adenomatous polyposis (FAP) and desmoid tumors.[48] Arthur Purdy Stout coined the term fibromatosis (in the name congenital generalized fibromatosis, describing myofibromatosis) in 1954.[49]
ICD-10-CM diagnosis codes
[edit]Few rare diseases have a specific code in the International Classification of Diseases.[50] As of October 2023, specific codes for desmoid tumors will be included in the ICD-10-CM, the United States' diagnosis code system, after a request from the Desmoid Tumor Research Foundation.[51] A subcategory of D48.1, Neoplasm of uncertain behavior of connective and other soft tissue, has been created with more specific codes:[50]
- D48.11: Desmoid tumor
- D48.110: Desmoid tumor of head and neck
- D48.111: Desmoid tumor of chest wall
- D48.112: Desmoid tumor, intrathoracic
- D48.113: Desmoid tumor of abdominal wall
- D48.114: Desmoid tumor, intraabdominal
- Desmoid tumor of pelvic cavity
- Desmoid tumor, peritoneal, retroperitoneal
- D48.115: Desmoid tumor of upper extremity and shoulder girdle
- D48.116: Desmoid tumor of lower extremity and pelvic girdle
- Desmoid tumor of buttock
- D48.117: Desmoid tumor of back
- D48.118: Desmoid tumor of other site
- D48.119: Desmoid tumor of unspecified site
Notable patients
[edit]- Dave Dravecky, American baseball pitcher and motivational speaker[52][53]
- Kevin Reilly, American football player[54]
In animals
[edit]Desmoid tumors occur in dogs, primarily on the head, and more infrequently in horses and cats.[55] A case has also been observed in a goat.[56]
References
[edit]- ^ a b c d e Master SR, Mangla A, Puckett Y, Shah C (January 2023). "Desmoid Tumor". StatPearls. Treasure Island: StatPearls Publishing. PMID 29083753. Archived from the original on 7 June 2023. Retrieved 14 August 2023 – via National Institutes of Health.
- ^ a b c d e f g Kasper B, Baumgarten C, Garcia J, Bonvalot S, Haas R, Haller F, et al. (October 2017). "An update on the management of sporadic desmoid-type fibromatosis: a European Consensus Initiative between Sarcoma PAtients EuroNet (SPAEN) and European Organization for Research and Treatment of Cancer (EORTC)/Soft Tissue and Bone Sarcoma Group (STBSG)". Annals of Oncology. 28 (10): 2399–2408. doi:10.1093/annonc/mdx323. PMC 5834048. PMID 28961825.
- ^ Nieuwenhuis MH, De Vos Tot Nederveen Cappel W, Botma A, Nagengast FM, Kleibeuker JH, Mathus-Vliegen EM, et al. (February 2008). "Desmoid tumors in a dutch cohort of patients with familial adenomatous polyposis". Clinical Gastroenterology and Hepatology. 6 (2): 215–219. doi:10.1016/j.cgh.2007.11.011. PMID 18237870. S2CID 26052046.
- ^ Sbaraglia M, Bellan E, Dei Tos AP (April 2021). "The 2020 WHO Classification of Soft Tissue Tumours: news and perspectives". Pathologica. 113 (2): 70–84. doi:10.32074/1591-951X-213. PMC 8167394. PMID 33179614.
- ^ "desmoid" at Dorland's Medical Dictionary
- ^ Lynch HT, Fitzgibbons R (December 1996). "Surgery, desmoid tumors, and familial adenomatous polyposis: case report and literature review". The American Journal of Gastroenterology. 91 (12): 2598–2601. PMID 8946994.
- ^ a b Valesano JC, Schmitz JJ, Jensen NM, Schultz GR, Callstrom MR (December 2017). "Desmoid Tumors: A Review of Their Natural History, Imaging, and Treatment". Journal of Radiology Nursing. 36 (4): 211–217. doi:10.1016/j.jradnu.2017.09.003.
- ^ a b c d e Howard JH, Pollock RE (June 2016). "Intra-Abdominal and Abdominal Wall Desmoid Fibromatosis". Oncology and Therapy. 4 (1): 57–72. doi:10.1007/s40487-016-0017-z. PMC 5315078. PMID 28261640.
- ^ a b "Desmoid tumor: MedlinePlus Genetics". National Library of Medicine MedlinePlus. Archived from the original on 26 October 2021. Retrieved 14 August 2023.
- ^ a b c d e f Kasper B, Ströbel P, Hohenberger P (May 2011). "Desmoid tumors: clinical features and treatment options for advanced disease". The Oncologist. 16 (5): 682–693. doi:10.1634/theoncologist.2010-0281. PMC 3228186. PMID 21478276.
- ^ a b c d e Weiss SW, Goldblum JR, Folpe AL (2007). "Deep Fibromatosis". Enzinger and Weiss's Soft Tissue Tumors (5th ed.). Elsevier Health Sciences. ISBN 9780323076159. Archived from the original on 14 August 2023. Retrieved 14 August 2023.
- ^ Sinha A, Tekkis PP, Gibbons DC, Phillips RK, Clark SK (November 2011). "Risk factors predicting desmoid occurrence in patients with familial adenomatous polyposis: a meta-analysis". Colorectal Disease. 13 (11): 1222–1229. doi:10.1111/j.1463-1318.2010.02345.x. PMID 20528895. S2CID 26117431.
- ^ Le Guellec S, Soubeyran I, Rochaix P, Filleron T, Neuville A, Hostein I, et al. (December 2012). "CTNNB1 mutation analysis is a useful tool for the diagnosis of desmoid tumors: a study of 260 desmoid tumors and 191 potential morphologic mimics". Modern Pathology. 25 (12): 1551–1558. doi:10.1038/modpathol.2012.115. PMID 22766794. S2CID 24471719.
- ^ Goldstein JA, Cates JM (July 2015). "Differential diagnostic considerations of desmoid-type fibromatosis". Advances in Anatomic Pathology. 22 (4): 260–266. doi:10.1097/PAP.0000000000000077. PMID 26050263. S2CID 3388912.
- ^ "Differential Diagnosis - Abdominal Desmoid Fibromatosis - Surgical Pathology Criteria". Stanford University School of Medicine. Archived from the original on 19 August 2023. Retrieved 19 August 2023.
- ^ a b c Kangas-Dick A, Ali M, Poss M, Khoury T, Takabe K (June 2024). "Diagnosis and Management of Desmoid Fibromatosis of the Breast". World Journal of Oncology. 15 (3): 394–404. doi:10.14740/wjon1844. ISSN 1920-4531. PMC 11092408. PMID 38751692.
- ^ a b c d e f g Bektas M, Bell T, Khan S, Tumminello B, Fernandez MM, Heyes C, et al. (September 2023). "Desmoid Tumors: A Comprehensive Review". Advances in Therapy. 40 (9): 3697–3722. doi:10.1007/s12325-023-02592-0. PMC 10427533. PMID 37436594.
- ^ a b c Peng PD, Hyder O, Mavros MN, Turley R, Groeschl R, Firoozmand A, et al. (December 2012). "Management and recurrence patterns of desmoids tumors: a multi-institutional analysis of 211 patients". Annals of Surgical Oncology. 19 (13): 4036–4042. doi:10.1245/s10434-012-2634-6. PMC 3568525. PMID 22972507.
- ^ "Desmoid Tumor - Symptoms, Causes, Treatment". National Organization for Rare Disorders. Archived from the original on 7 May 2023. Retrieved 7 May 2023.
- ^ a b c Lorenzen J, Cramer M, Buck N, Friedrichs K, Graubner K, Lühr CS, et al. (February 2021). "Desmoid Type Fibromatosis of the Breast: Ten-Year Institutional Results of Imaging, Histopathology, and Surgery". Breast Care. 16 (1): 77–84. doi:10.1159/000507842. PMC 7923936. PMID 33708054.
- ^ "Desmoid Tumor - Risk Factors". Cancer.Net. American Society of Clinical Oncology. 2 September 2020. Archived from the original on 19 August 2023. Retrieved 19 August 2023.
- ^ a b c "Desmoid Tumor - Statistics". Cancer.Net. American Society of Clinical Oncology. 2 September 2020. Archived from the original on 14 August 2023. Retrieved 14 August 2023.
- ^ a b c Alman B, Attia S, Baumgarten C, Benson C, Blay JY, Bonvalot S, et al. (Desmoid Tumor Working Group) (March 2020). "The management of desmoid tumours: A joint global consensus-based guideline approach for adult and paediatric patients". European Journal of Cancer. 127: 96–107. doi:10.1016/j.ejca.2019.11.013. hdl:2318/1793788. PMID 32004793. S2CID 210998595.
- ^ Sinha A, Tekkis PP, Neale KF, Phillips RK, Clark SK (June 2010). "Risk factors predicting intra-abdominal desmoids in familial adenomatous polyposis: a single centre experience". Techniques in Coloproctology. 14 (2): 141–146. doi:10.1007/s10151-010-0573-4. PMID 20352275. S2CID 24922322.
- ^ Sinha A, Gibbons DC, Phillips RK, Clark S (September 2010). "Surgical prophylaxis in familial adenomatous polyposis: do pre-existing desmoids outside the abdominal cavity matter?". Familial Cancer. 9 (3): 407–411. doi:10.1007/s10689-010-9342-9. PMID 20428953. S2CID 20685381.
- ^ a b Li GZ, Raut CP, Hunt KK, Feng M, Chugh R (March 2021). "Breast Sarcomas, Phyllodes Tumors, and Desmoid Tumors: Epidemiology, Diagnosis, Staging, and Histology-Specific Management Considerations". American Society of Clinical Oncology Educational Book. American Society of Clinical Oncology. Annual Meeting. 41 (41): 390–404. doi:10.1200/EDBK_321341. PMID 34010054.
- ^ Rammohan A, Wood JJ (2012). "Desmoid tumour of the breast as a manifestation of Gardner's syndrome". International Journal of Surgery Case Reports. 3 (5): 139–142. doi:10.1016/j.ijscr.2012.01.004. PMC 3312056. PMID 22370045.
- ^ Yalla P, Rathod P, Rojesara M, Pawar A, Devarajan JA, Pandya SJ (December 2023). "A Spindle Cell Tumour that Took us for a Spin!: A Case Report and Short Review of Management of Head and Neck Desmoid Fibromatosis". Indian Journal of Otolaryngology and Head & Neck Surgery. 75 (4): 4028–4031. doi:10.1007/s12070-023-04008-5. ISSN 2231-3796. PMC 10646034. PMID 37974872.
- ^ a b "FDA Approves First Therapy for Rare Type of Non-Cancerous Tumors". U.S. Food and Drug Administration (FDA) (Press release). 27 November 2023. Archived from the original on 28 November 2023. Retrieved 28 November 2023.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain.
- ^ "SpringWorks Therapeutics Announces FDA Approval of Ogsiveo (nirogacestat) as the First and Only Treatment for Adults with Desmoid Tumors" (Press release). SpringWorks Therapeutics. 27 November 2023. Archived from the original on 28 November 2023. Retrieved 28 November 2023 – via GlobeNewswire.
- ^ "A Study of AL102 in Patients With Progressing Desmoid Tumors (RINGSIDE)". ClinicalTrials.gov. Retrieved 19 August 2023.
- ^ Sava J (7 November 2023). "FDA Grants Orphan Drug Designation to AL102 in Desmoid Tumors". Targeted Oncology. Retrieved 4 December 2023.
- ^ a b Mangla A, Agarwal N, Schwartz G (25 January 2024). "Desmoid Tumors: Current Perspective and Treatment". Current Treatment Options in Oncology. 25 (2): 161–175. doi:10.1007/s11864-024-01177-5. ISSN 1527-2729. PMC 10873447. PMID 38270798.
- ^ McLean TD, Duchi S, Di Bella C (May 2022). "Molecular Pathogenesis of Sporadic Desmoid Tumours and Its Implications for Novel Therapies: A Systematised Narrative Review". Targeted Oncology. 17 (3): 223–252. doi:10.1007/s11523-022-00876-z. ISSN 1776-2596. PMC 9217905. PMID 35446005.
- ^ a b Fernandez MM, Bell T, Tumminello B, Khan S, Zhou S, Oton AB (3 July 2023). "Disease and economic burden of surgery in desmoid tumors: a review". Expert Review of Pharmacoeconomics & Outcomes Research. 23 (6): 607–618. doi:10.1080/14737167.2023.2203915. PMID 37099290.
- ^ Ganeshan D, Amini B, Nikolaidis P, Assing M, Vikram R (2019). "Current Update on Desmoid Fibromatosis". Journal of Computer Assisted Tomography. 43 (1): 29–38. doi:10.1097/RCT.0000000000000790. PMC 6331223. PMID 30211798.
- ^ "Diagnosis and Treatment". Desmoid Tumor Research Foundation. Archived from the original on 25 October 2021. Retrieved 22 October 2021.
- ^ a b Lee YS, Joo MW, Shin SH, Hong S, Chung YG (8 January 2024). "Current Treatment Concepts for Extra-Abdominal Desmoid-Type Fibromatosis: A Narrative Review". Cancers. 16 (2): 273. doi:10.3390/cancers16020273. ISSN 2072-6694. PMC 10813957. PMID 38254764.
- ^ Chatzipetrou MA, Tzakis AG, Pinna AD, Kato T, Misiakos EP, Tsaroucha AK, et al. (March 2001). "Intestinal transplantation for the treatment of desmoid tumors associated with familial adenomatous polyposis". Surgery. 129 (3): 277–281. doi:10.1067/msy.2001.110770. PMID 11231455.
- ^ Schwartz RA, Lambert PC, Shear N (22 June 2023). Butler DF, Elston DM (eds.). "Desmoid Tumor: Practice Essentials, Pathophysiology, Etiology". eMedicine. Archived from the original on 18 September 2023. Retrieved 14 August 2023.
- ^ a b c d e f g h Kasper B, Gounder M, Hernandez L, Baumgarten C, Ratan R (7 June 2024). "Capturing Patient Voice to Improve Outcomes That Matter to Patients with Desmoid Tumor". Cancer Management and Research. 16: 617–628. doi:10.2147/CMAR.S362694. PMC 11166168. PMID 38863992.
- ^ Wilkinson MJ, Fitzgerald JE, Thomas JM, Hayes AJ, Strauss DC (May 2012). "Surgical resection for non-familial adenomatous polyposis-related intra-abdominal fibromatosis". The British Journal of Surgery. 99 (5): 706–713. doi:10.1002/bjs.8703. PMID 22359346. S2CID 205512855.
- ^ a b Husson O, Younger E, Dunlop A, Dean L, Strauss DC, Benson C, et al. (March 2019). "Desmoid fibromatosis through the patients' eyes: time to change the focus and organisation of care?". Supportive Care in Cancer. 27 (3): 965–980. doi:10.1007/s00520-018-4386-8. PMC 6373240. PMID 30155568.
- ^ a b Garg V, Rastogi S, Kalra K, Bhoriwal S, Barwad A, Dhamija E, et al. (December 2022). "Health-related quality of life (HRQoL), anxiety, and depression in patients with desmoid type fibromatosis". Supportive Care in Cancer. 30 (12): 10089–10098. doi:10.1007/s00520-022-07445-0. PMID 36350382. S2CID 253418898.
- ^ a b Ravi V, Patel SR, Raut CP, Baldini EH, Berman RS, Pollock RE (January 2022). "Desmoid tumors: Epidemiology, molecular pathogenesis, clinical presentation, diagnosis, and local therapy". UpToDate. Archived from the original on 14 August 2023. Retrieved 14 August 2023.
- ^ Hajdu SI (May 2007). "Soft tissue sarcomas". Cancer. 109 (9): 1697–1704. doi:10.1002/cncr.22608. PMID 17366588. S2CID 39827598.
- ^ a b Nichols RW (1 July 1923). "Desmoid tumors: a report of thirty-one cases". Archives of Surgery. 7 (1): 227. doi:10.1001/archsurg.1923.01120010230013. ISSN 0004-0010.
- ^ "Gardner Syndrome: Practice Essentials, Anatomy, Pathophysiology". eMedicine. 29 April 2022. Archived from the original on 14 August 2023. Retrieved 14 August 2023.
- ^ Beck JC, Devaney KO, Weatherly RA, Koopmann CF, Lesperance MM (January 1999). "Pediatric myofibromatosis of the head and neck". Archives of Otolaryngology–Head & Neck Surgery. 125 (1): 39–44. doi:10.1001/archotol.125.1.39. PMID 9932585. S2CID 19440724.
- ^ a b "Diagnosis Codes & Desmoid Tumors". Desmoid Tumor Research Foundation. Archived from the original on 12 August 2023. Retrieved 12 August 2023.
- ^ "ICD-10 Coordination and Maintenance Committee Meeting" (PDF). Centers for Disease Control and Prevention. 8–9 March 2022. Archived (PDF) from the original on 15 May 2022. Retrieved 12 August 2023.
- ^ Nack W (22 July 1991). "'Let's Make the Best of It'". Sports Illustrated Vault. Archived from the original on 9 July 2023. Retrieved 14 August 2023.
- ^ "Former MLB player Dave Dravecky to Headline Cancer Survivorship Conference Sept. 16-17". MD Anderson Cancer Center. 14 June 2011. Archived from the original on 14 August 2023. Retrieved 14 August 2023.
- ^ "Kevin Reilly - Sarcoma Cancer Research & Treatment". Sarcoma Foundation of America. Retrieved 16 August 2023.
- ^ "Connective Tissue Tumors in Animals - Integumentary System". MSD Veterinary Manual. Archived from the original on 14 August 2023. Retrieved 14 August 2023.
- ^ Tontis A, Rossi GL (August 1993). "[First description of a desmoid tumor in a goat and comparative observations to human fibromatosis. Case report]". Tierärztliche Praxis (in German). 21 (4): 306–311. PMID 8211956.
Aggressive fibromatosis
View on GrokipediaIntroduction and Background
Definition and characteristics
Aggressive fibromatosis, also known as desmoid tumor, desmoid-type fibromatosis, or deep fibromatosis, is a rare clonal fibroblastic proliferation of myofibroblasts that forms a locally invasive soft tissue tumor without metastatic potential.[1][6] It arises from deep soft tissues, including fascia, musculoaponeurotic structures, and periosteum, and is characterized by infiltrative growth that can encroach on adjacent structures such as muscles, nerves, and vessels.[7] Despite its benign histology, the tumor exhibits aggressive local behavior, with a notable risk of recurrence following incomplete excision, often ranging from 20% to 30%.[6] Microscopically, aggressive fibromatosis consists of uniform spindle-shaped fibroblastic or myofibroblastic cells arranged in long, sweeping fascicles within a collagenous stroma. These cells display pale cytoplasm, minimal nuclear atypia, and lack significant hyperchromasia, with variable collagen deposition that may include areas of hyalinization or myxoid change.[6] The stroma often features thin-walled vessels and occasional microhemorrhages, while the overall architecture shows no necrosis and a typically low mitotic rate.[6] It is sometimes associated with genetic mutations, such as those in the APC or CTNNB1 genes, though these are explored in greater detail elsewhere.[8] This entity is distinguished from sarcomas by its absence of malignant features, including high mitotic activity, marked cellular pleomorphism, and necrosis, which are hallmarks of true soft tissue malignancies.[6] Instead, aggressive fibromatosis maintains a bland cytologic appearance and clonal but non-metastasizing proliferation, underscoring its intermediate behavior between benign fibromatoses and sarcomas.[1]History and etymology
The first description of what is now known as aggressive fibromatosis dates to 1832, when Scottish surgeon John Macfarlane reported a tendon-like fibrous tumor arising from the abdominal wall of a 30-year-old woman during a postmortem examination.[9] In 1838, German pathologist Johannes Peter Müller coined the term "desmoid" for these lesions, derived from the Greek word desmos, meaning "band" or "tendon," to reflect their firm, band-like consistency and resemblance to tendinous tissue.[10] During the 19th century, desmoid tumors were often misclassified as fibrosarcomas due to their infiltrative growth pattern and histological similarity to malignant fibrous neoplasms, leading to aggressive surgical interventions despite their lack of metastatic potential.[11] By the mid-20th century, pathologists began reclassifying them to emphasize their benign but locally invasive nature; the term "fibromatosis" was introduced by Arthur Purdy Stout in the 1940s and 1950s to describe a spectrum of fibroblastic proliferations, including desmoids, distinguishing them from true sarcomas.[12] This shift culminated in the widespread adoption of "aggressive fibromatosis" as a descriptor in the late 20th century, highlighting their non-metastasizing yet recurrence-prone behavior without implying outright malignancy.[10] Key milestones in understanding aggressive fibromatosis include Stout's 1948 seminal paper on fibrosarcomas, which helped delineate desmoids as a distinct entity through detailed histopathological analysis.[13] Additionally, the association between desmoid tumors and familial adenomatous polyposis (FAP) was established in the mid-20th century through the description of Gardner syndrome, an inherited condition caused by APC gene mutations, prompting targeted screening and management strategies for at-risk patients.[14] Today, aggressive fibromatosis is classified by the World Health Organization as an intermediate fibroblastic and myofibroblastic tumor.[6]Epidemiology and Occurrence
Incidence and demographics
Aggressive fibromatosis, also known as desmoid tumor, is a rare soft tissue neoplasm with an estimated annual incidence of 2 to 5 cases per 1,000,000 people worldwide.[2][1][15] It accounts for approximately 0.03% of all tumors and less than 3% of soft tissue tumors.[2][15] The condition predominantly affects individuals between 15 and 60 years of age, with the highest frequency observed in the 20- to 44-year-old age group; it is rare in children younger than 5 years and in adults over 70.[16][1] There is a notable female predominance, with a sex ratio ranging from 2:1 to 3:1, potentially influenced by hormonal factors such as estrogen.[2][15] Geographically, aggressive fibromatosis shows no strong ethnic or regional variations, though higher reported rates in Western populations may reflect improved diagnostic capabilities rather than true prevalence differences.[15] In patients with familial adenomatous polyposis (FAP), the lifetime risk of developing the tumor is substantially elevated, reaching 20% to 30%.[2][15]Anatomic locations and variants
Aggressive fibromatosis, also known as desmoid-type fibromatosis, primarily arises in three anatomic categories based on location: extra-abdominal, abdominal wall, and intra-abdominal, with distinct distributions and clinical implications. Abdominal wall tumors account for approximately 50% of cases and frequently develop in women, often associated with postpartum changes or prior surgical scars like those from cesarean sections.[1][17] Extra-abdominal tumors most commonly occur in the extremities (such as the shoulder and thigh), trunk, and head/neck regions and comprise about 40% of cases; these are typically sporadic and involve deep musculoaponeurotic structures.[1][18] Intra-abdominal tumors comprise about 10% of cases and predominantly involve the mesentery or retroperitoneum; these are more frequently linked to familial adenomatous polyposis (FAP) and exhibit heightened local aggressiveness.[1][18] Key variants of aggressive fibromatosis include sporadic forms, which constitute 85-90% of cases driven by somatic CTNNB1 mutations, versus FAP-related forms (5-10%) associated with germline APC mutations, with the latter more prevalent in intra-abdominal sites.[1] Additionally, while superficial fibromatoses (such as palmar or plantar types) are genetically distinct and generally less aggressive with smaller size and lower recurrence, aggressive fibromatosis itself is classified as the deep musculoaponeurotic subtype, which is rare in superficial locations and characterized by infiltrative growth into muscle and fascia.[19] Site-specific behaviors vary significantly; tumors in the extremities often cause symptoms due to mechanical compression, leading to pain and restricted mobility, whereas intra-abdominal lesions in the mesentery or retroperitoneum carry a substantial risk of bowel obstruction and higher recurrence rates in FAP-associated cases (75-85%).[20] Abdominal wall variants, particularly in women, may show accelerated growth during or after pregnancy but generally have lower recurrence following resection compared to deeper sites.[17]Etiology and Pathogenesis
Risk factors and triggers
Aggressive fibromatosis, also known as desmoid tumor, exhibits several non-genetic risk factors that predispose individuals to its development, including female sex and young adulthood as demographic amplifiers. The condition occurs more frequently in women, with a female-to-male ratio of approximately 2:1, particularly during reproductive years. Incidence peaks in young adults aged 20 to 40 years, though it can arise at any age.[1][2] Hormonal influences, particularly estrogen exposure, play a notable role in triggering the tumor's onset and progression. Higher incidence is observed during or shortly after pregnancy, with some cases linked to abdominal wall desmoids emerging postpartum, potentially due to elevated estrogen levels and tissue changes. Estrogen's involvement is further suggested by tumor regression during menopause and associations with oral contraceptive use, as well as potential fluctuations tied to the menstrual cycle.[1][4][2] Trauma and prior surgery represent key local triggers, especially in sporadic cases comprising the majority of occurrences. Approximately 10-20% of sporadic aggressive fibromatosis cases arise at sites of previous surgical scars, such as those from cesarean sections or other abdominal procedures, with a median lag time of about 23 months post-trauma. Physical trauma, including blunt injuries, is implicated in around 7-20% of cases, often leading to tumor development at the injury site. Prior radiation exposure is a rare trigger, reported in isolated instances but not strongly established as a common risk. In contrast, no robust links exist between tobacco use, alcohol consumption, or these tumors.[21][1] Approximately 5-10% of aggressive fibromatosis cases are associated with familial syndromes, notably familial adenomatous polyposis (FAP) or Gardner syndrome, where tumors predominate in the abdomen and often emerge post-surgery. These cases stem from underlying genetic underpinnings in FAP, such as APC gene alterations, which amplify susceptibility but are detailed in molecular mechanisms.[1][2]Genetic and molecular mechanisms
Aggressive fibromatosis, also known as desmoid-type fibromatosis, primarily arises through dysregulation of the Wnt/β-catenin signaling pathway, driven by specific genetic alterations in most cases.[22] Sporadic cases, comprising 85-90% of all instances, are characterized by somatic mutations in the CTNNB1 gene, which encodes β-catenin, occurring in 70-85% of tumors. Among CTNNB1 mutations, S45F is associated with higher recurrence risk compared to T41A.[1] These mutations cluster in exon 3 and typically produce missense substitutions at codons 41 or 45, including T41A (threonine to alanine), S45F (serine to phenylalanine), and S45P (serine to proline), with T41A being the most prevalent at approximately 50-60% of mutated cases, followed by S45F (30-35%) and S45P (around 8%).[23][22] In addition, somatic APC gene mutations or loss-of-function alterations are detected in approximately 3-5% of sporadic tumors, further contributing to pathway activation.[22][24] In contrast, the 5-10% of cases associated with familial adenomatous polyposis (FAP) feature germline APC mutations, which truncate the APC protein and impair its role in the β-catenin destruction complex, leading to constitutive Wnt pathway dysregulation.[25][20] These mutations stabilize β-catenin by preventing its ubiquitination and proteasomal degradation, resulting in its nuclear translocation and accumulation—a hallmark feature observed in over 95% of aggressive fibromatosis tumors regardless of etiology.[23][22] Aberrant Wnt/β-catenin signaling promotes myofibroblastic proliferation through transcriptional activation of target genes, including c-Myc, which regulates cell growth and survival.[26] Mutations in either CTNNB1 or APC disrupt the destruction complex (comprising APC, AXIN, GSK3β, and CK1), halting β-catenin phosphorylation at key serine/threonine residues and enabling its binding to TCF/LEF transcription factors.[22][27] While rare alterations in other Wnt regulators such as LRP6 or AXIN1 have been implicated in isolated cases, no consistent chromosomal abnormalities, such as recurrent translocations or aneuploidy, are identified across the disease spectrum.[24][22]Clinical Features
Signs and symptoms
Aggressive fibromatosis, also known as desmoid tumor, most commonly presents as a painless, firm, slow-growing mass that may be discovered incidentally during routine imaging or physical examination for unrelated issues.[4][28] Many cases are asymptomatic at diagnosis, particularly smaller extra-abdominal tumors, allowing for initial observation without immediate intervention.[1][29] Symptoms vary significantly by anatomic location due to the tumor's local invasiveness. In the extremities, patients often experience pain, swelling, and limited range of motion as the mass compresses surrounding muscles, nerves, or joints. Abdominal wall tumors typically manifest as a palpable lump, sometimes following prior trauma or surgery in the area. Intra-abdominal desmoid tumors can cause abdominal pain, cramping, nausea, or more severe complications such as bowel obstruction and hydronephrosis from ureteral compression.[30][4][31] In advanced cases, the tumor's growth leads to functional impairments, including neuropathy from nerve compression or reduced mobility in affected limbs. Rare systemic effects, such as weight loss, may occur if intra-abdominal tumors interfere with digestion or cause chronic malaise. Progression is generally gradual, with enlargement occurring over months to years, though rare instances of rapid growth have been reported.[1][32][33]Natural disease course
Aggressive fibromatosis, also known as desmoid-type fibromatosis, exhibits a highly variable natural history characterized by local invasiveness without metastatic potential. The tumor typically demonstrates indolent behavior, infiltrating surrounding structures such as muscle, bone, and viscera, but it does not spread distantly.[3][34] The growth patterns of untreated tumors are unpredictable, with approximately 50-60% remaining stable after diagnosis, showing no significant enlargement over periods of at least 6 months to several years. In observational studies, up to 70% of cases display slow or no progression, while a subset may exhibit initial growth followed by stabilization. Spontaneous regression occurs in 10-30% of cases, particularly in abdominal wall tumors associated with pregnancy, where postpartum resolution has been documented in up to 30% of instances.[34][35][3] Without intervention, the disease follows a chronic trajectory, with median time to progression estimated at 1-2 years in progressing cases, though many enter a prolonged stable phase lasting years. Local regrowth or progression after apparent stability is possible but uncommon, and malignant transformation is exceedingly rare, with no distant metastasis reported in natural history cohorts.[34][35][36] Factors influencing the course include tumor location and genetic associations; extra-abdominal lesions often follow a more predictable, less aggressive pattern compared to intra-abdominal ones, while those linked to familial adenomatous polyposis (FAP) tend to be more proliferative and less prone to spontaneous regression.[3][34]Diagnosis
Diagnostic approaches
Diagnosis of aggressive fibromatosis, also known as desmoid-type fibromatosis, relies on a combination of imaging modalities and histopathological confirmation to distinguish it from other soft tissue tumors.[1] Imaging plays a crucial role in initial evaluation, with magnetic resonance imaging (MRI) considered the gold standard for assessing lesion extent, infiltration, and surgical planning due to its superior soft tissue contrast. On MRI, lesions typically appear isointense to skeletal muscle on T1-weighted images and heterogeneously hyperintense on T2-weighted images, often exhibiting a "band sign" of low-signal intensity bands representing dense collagen bundles. Post-contrast enhancement is moderate to marked in approximately 90% of cases, correlating with higher T2 signal intensity and more aggressive growth patterns. Advanced techniques like diffusion-weighted MRI enhance lesion characterization. Computed tomography (CT) is useful for detecting calcifications, particularly in intra-abdominal or mesenteric lesions, where tumors may show ill-defined margins and a whorled appearance, though it is less sensitive for soft tissue delineation than MRI. Ultrasound serves as a first-line tool for superficial, palpable lesions, revealing oval solid masses with variable echogenicity, smooth or ill-defined margins, and heterogeneous vascularity on color Doppler, aiding in biopsy guidance.[37][38][1] Definitive diagnosis requires tissue sampling via biopsy, as imaging alone cannot reliably exclude malignancy. Core needle biopsy, often imaging-guided, is preferred for its adequacy in providing sufficient material for histopathological analysis, while incisional biopsy may be used for larger or deeper lesions to ensure representative sampling. Excisional biopsy is generally avoided initially to prevent potential seeding along the biopsy tract in infiltrative tumors. Molecular testing for CTNNB1 or APC mutations is increasingly routine for confirming diagnosis and assessing recurrence risk, as recommended in current guidelines.[6][1] Histopathological examination reveals a proliferation of uniform spindle-shaped myofibroblasts arranged in sweeping fascicles or a storiform pattern within a collagenous stroma, with minimal nuclear atypia, low mitotic activity, and infiltration of surrounding tissues without encapsulation. Immunohistochemistry is confirmatory, showing nuclear positivity for beta-catenin in over 90% of cases due to CTNNB1 mutations, cytoplasmic positivity for smooth muscle actin (SMA), and negativity for desmin and S-100 protein, helping to rule out myogenic or neural tumors. Genetic testing for CTNNB1 or APC mutations may support the diagnosis in select cases, particularly those associated with familial adenomatous polyposis.[6][1] Differential diagnosis includes sarcomas (e.g., low-grade fibrosarcoma), reactive processes (e.g., nodular fasciitis), and other fibroproliferative lesions (e.g., gastrointestinal stromal tumors or endometriosis in abdominal sites), which are excluded through the combination of imaging characteristics, histopathologic features lacking necrosis or high mitotic rates, and specific IHC profiles. A multidisciplinary approach involving radiologists, pathologists, and surgeons is essential for integrated assessment and accurate diagnosis.[37][1][6]Classification systems
Aggressive fibromatosis, also known as desmoid-type fibromatosis, is classified by the World Health Organization (WHO) under the category of fibroblastic and myofibroblastic tumors as an intermediate (locally aggressive) neoplasm.[39] This designation reflects its infiltrative growth pattern and tendency for local recurrence without metastatic potential, distinguishing it from both benign fibrous lesions and high-grade sarcomas.[40] The 2020 WHO classification emphasizes its clonal fibroblastic proliferation arising in deep soft tissues.[41] Classification systems also categorize cases based on anatomic location, which influences clinical presentation and management. Extra-abdominal desmoids typically occur in the extremities, trunk, head, or neck, accounting for 40-50% of cases and often presenting as painless masses.[41] Abdominal wall fibromatosis, comprising about 25-35% of occurrences, is frequently associated with prior trauma or pregnancy in women of childbearing age.[6] Intra-abdominal or mesenteric variants, representing 15-20%, arise within the peritoneal cavity and are more common in syndromic contexts, potentially causing bowel obstruction or vascular compression.[42] Etiologic subtypes further refine classification into sporadic and syndromic forms. Sporadic desmoid-type fibromatosis, which constitutes 85-90% of cases, is primarily driven by somatic mutations in the CTNNB1 gene encoding beta-catenin.[41] In contrast, syndromic cases (10-15%) are linked to germline mutations in the APC gene, most notably in familial adenomatous polyposis (FAP), where desmoids often develop intra-abdominally and contribute significantly to morbidity.[43] Historically, aggressive fibromatosis was misclassified as a low-grade fibrosarcoma (grade 1) due to its infiltrative behavior, leading to overly aggressive treatments in the mid-20th century.[1] By the 1980s, evolving histopathological understanding reclassified it as a non-malignant, borderline tumor, shifting focus from radical resection to more conservative approaches and recognizing its lack of metastatic capability.[44] Prognostic subclassification within sporadic cases often incorporates beta-catenin mutation types, as specific CTNNB1 variants correlate with recurrence risk post-resection. For instance, the S45F mutation is associated with a higher likelihood of local recurrence (up to 70-80% in some cohorts) compared to T41A or wild-type tumors, guiding risk-adapted surveillance.[45] This molecular stratification, while not universal, aids in identifying higher-risk subsets without altering the intermediate-grade status.[46]Staging and risk stratification
Aggressive fibromatosis, also known as desmoid tumor, lacks a formal TNM staging system due to its non-metastatic nature, relying instead on clinical risk models that incorporate tumor site, size, symptoms, and patient factors to predict progression and recurrence.[47] These models guide management by identifying low-risk cases suitable for observation versus high-risk cases requiring intervention.[48] The Memorial Sloan Kettering Cancer Center (MSKCC) nomogram represents a widely used tool for estimating local recurrence-free survival after surgery, based on factors such as age, tumor site (e.g., extremity versus abdominal), size, and margin status.[49] In this system, low-risk features include extremity location, tumor size less than 5 cm, and asymptomatic presentation, while high-risk characteristics encompass intra-abdominal site, size greater than 10 cm, and symptomatic disease, with extremity tumors and sizes over 5 cm independently associated with higher relapse rates.[48] Younger age (under 30 years) further elevates risk across categories.[47] For cases associated with familial adenomatous polyposis (FAP), staging focuses on intra-abdominal tumors and assesses multiplicity, invasion, and clinical impact, distinguishing unicentric (solitary) from multicentric (multiple) lesions that infiltrate critical structures like the mesentery or vasculature.[50] A dedicated staging system classifies these into four stages: Stage I (small, asymptomatic, unicentric tumors without complications); Stage II (moderate size or symptoms, limited invasion); Stage III (larger, symptomatic with significant obstruction or invasion); and Stage IV (extensive multicentric disease causing severe complications like bowel obstruction or ureteral involvement).[51] In a 2012 study of 154 patients, 5-year mortality was 0% for stages I and II, 11% for stage III, and 24% for stage IV due to complications.[52] This FAP-specific approach highlights poorer outcomes in advanced stages.[51] Key risk factors for progression include positive surgical margins, young age at diagnosis, and trunk or abdominal location, which correlate with higher recurrence rates.[48] Histologic assessment, including tools evaluating mitotic activity and specific CTNNB1 mutations (e.g., S45F), aids in further stratifying risk, as certain mutations predict aggressive behavior.[53] These stratification methods inform decisions between watchful waiting for low-risk disease and active therapy for high-risk cases, with 5-year progression-free survival ranging from 50% in high-risk groups to 80% in low-risk cohorts.[49]Management and Treatment
Therapeutic options
The management of aggressive fibromatosis, also known as desmoid-type fibromatosis, emphasizes a conservative, individualized strategy that prioritizes function preservation and minimizes morbidity, guided by multidisciplinary tumor boards involving surgeons, oncologists, radiologists, and pathologists.[54] Treatment selection is risk-based, considering tumor site, symptoms, growth kinetics, and patient factors such as age and comorbidities, with active surveillance often preferred for low-risk cases.[55] Watchful waiting, or active surveillance, is the frontline approach for asymptomatic or minimally symptomatic tumors, particularly those in critical locations like the mesentery or retroperitoneum, where intervention risks outweigh benefits.[54] This involves serial imaging with MRI (preferred for soft tissue evaluation) or CT every 3 months initially, extending to 6 months if stable, to monitor for progression, which occurs in approximately 50% of cases at 5 years, while 20-30% may spontaneously regress.[56] Evidence supports this strategy with level IV data and a grade B recommendation, as it avoids unnecessary treatments in indolent disease.[54] Surgery remains an option for symptomatic, progressive, or resectable tumors where complete excision is feasible with acceptable morbidity, such as isolated abdominal wall lesions.[55] The goal is R0 resection (negative margins) for optimal local control, achieving approximately 80% at 5 years, though function-sparing techniques are prioritized over aggressive margins due to high recurrence rates (25-60%) and potential complications like nerve damage or organ loss.[54] Morbidity concerns limit its use in intra-abdominal or extremity sites, with decisions deferred to expert consensus (level IV evidence, grade A recommendation).[56] Radiation therapy is reserved for inoperable, symptomatic, or recurrent tumors not amenable to surgery, delivering moderate doses of 50-56 Gy in 1.8-2 Gy fractions to achieve local control in 70-80% of progressive cases.[54] It may be used adjuvantly for positive margins but is approached cautiously due to risks of secondary malignancies (up to 5-10% lifetime) and fibrosis, particularly in young patients or radiosensitive sites (level III evidence, grade A recommendation).[55] Systemic medical therapy serves as a nonsurgical alternative for advanced, unresectable, or multi-site disease, starting with low-toxicity options like NSAIDs (e.g., sulindac) or anti-estrogens (e.g., tamoxifen) for first-line management of slowly progressive tumors, offering stabilization in many cases with minimal side effects (level III evidence, grade B recommendation).[54] For more aggressive or symptomatic cases, low-dose chemotherapy regimens such as methotrexate combined with vinblastine or vinorelbine provide disease control in 60-70% of patients, particularly in critical intra-abdominal locations, though reserved for progression due to potential myelosuppression (level III evidence, grade B recommendation).[55] Overall, therapeutic decisions follow NCCN and ESMO guidelines, advocating a tailored, multidisciplinary framework that integrates patient preferences and serial assessments to escalate from surveillance to intervention only when necessary.[56][54]Research and emerging therapies
Targeted therapies have emerged as a cornerstone of research in aggressive fibromatosis, particularly gamma-secretase inhibitors that disrupt the Notch signaling pathway implicated in tumor pathogenesis. Nirogacestat, an oral gamma-secretase inhibitor, received FDA approval in November 2023 for adult patients with progressing desmoid tumors requiring systemic treatment, based on the phase 3 DeFi trial demonstrating a 41% objective response rate compared to 8% with placebo and a progression-free survival hazard ratio of 0.29.[57][5] Nirogacestat also received European approval in August 2025 for adult patients with progressing desmoid tumors. This approval marked the first systemic therapy specifically for desmoid tumors, with long-term follow-up data from the DeFi trial presented in October 2025 showing durable objective responses, further tumor size reductions (median -75.8% at 4 years in long-term patients), median treatment duration of 33.6 months, and improved pain scores.[58][59][60] Tyrosine kinase inhibitors targeting the beta-catenin/Wnt pathway, central to aggressive fibromatosis biology, continue to show promise in clinical studies. Sorafenib, a multi-kinase inhibitor, achieved stable disease in approximately 70% of patients in the phase 3 Alliance A091105 trial, with 81% progression-free survival at 2 years versus 30% for placebo, confirming its role in delaying progression for unresectable or symptomatic cases.[61][62] Pazopanib, another tyrosine kinase inhibitor, demonstrated symptom control in 75% of patients and no radiological progression (100% stability) in a retrospective series of 8 patients, supporting its use in beta-catenin-driven tumors.[63] Imatinib, targeting PDGFR and c-KIT, has shown disease control rates around 68% in phase 2 trials, with 1-year progression-free survival of 66%.[64] Ongoing clinical trials are expanding access to these agents globally and exploring combinations for refractory disease. A phase 3 trial of nirogacestat in Japanese adults with desmoid tumors initiated in September 2025 aims to evaluate efficacy and safety in this population, building on DeFi results.[65] For refractory cases, interferon-alpha has shown partial responses in historical cohorts, while gefitinib achieved sustained partial response in a 2025 case report of an EGFR 19del-mutated intra-abdominal tumor after multiple prior therapies.[66] In pediatric settings, ultrasound-guided microwave ablation demonstrated feasibility and safety for recurrent aggressive fibromatosis, with complete response in 20% of lesions (2/10) and overall response in 80%, with no major complications in a 2025 prospective study of postoperative recurrences.[67] Recent advances from 2023 to 2025 emphasize health-related quality of life (HRQoL) improvements with targeted agents and the role of genetic profiling in personalization. The DeFi trial reported significant HRQoL gains with nirogacestat, including reduced pain interference and better physical functioning scores versus placebo.[68] Genetic profiling of CTNNB1 and APC mutations now guides therapy selection, with 2024 studies linking specific variants to imatinib sensitivity and supporting tailored approaches to avoid ineffective treatments.[69][70] Despite progress, challenges persist due to the high recurrence rate of 20-50% post-treatment and the need for de-escalation strategies to minimize overtreatment morbidity. Research in 2025 highlights active surveillance for stable tumors, reducing unnecessary interventions while monitoring for progression via imaging.[71][72]Prognosis and Patient Impacts
Prognostic factors
Aggressive fibromatosis, also known as desmoid tumor, exhibits a highly variable prognosis influenced by several clinical and molecular factors. Favorable prognostic indicators include the potential for spontaneous regression, which occurs in approximately 10-20% of cases, particularly in extra-abdominal locations.[73] Complete surgical resection with negative margins (R0) is associated with local control rates of 70-90%, significantly reducing the risk of recurrence.[74] Extra-abdominal sites, such as the extremities or trunk, generally portend better outcomes compared to intra-abdominal tumors, with lower rates of aggressive behavior and complications.[55] Unfavorable factors include intra-abdominal location and association with familial adenomatous polyposis (FAP), where recurrence rates can reach 50-70% or higher following treatment.[55] Young age at diagnosis, particularly under 30 years, correlates with increased recurrence risk due to more aggressive tumor biology.[75] Large tumor size exceeding 10 cm is linked to higher progression and poorer response to interventions.[55] Positive surgical margins (R1 or R2) substantially elevate the likelihood of local relapse, often necessitating additional therapies.[76] Disease-specific survival approaches 100%, as aggressive fibromatosis does not metastasize and remains histologically benign, though local invasion can lead to morbidity.[1] The 5-year progression-free survival rate typically ranges from 50-70%, varying by treatment modality and tumor characteristics.[77] Most recurrences occur within 24 months post-treatment. Multifocal disease is rare in sporadic cases.[78] Ongoing monitoring is essential and involves serial imaging, such as MRI every 3-6 months initially, to detect progression early.[76] Specific beta-catenin (CTNNB1) mutations, notably S45F, serve as a molecular biomarker for higher recurrence risk, guiding risk stratification and surveillance intensity.[1] Recent systemic therapies like nirogacestat (FDA-approved in November 2023) have demonstrated improved progression-free survival, potentially enhancing long-term prognosis.[79]Physical and psychosocial impacts
Aggressive fibromatosis, also known as desmoid tumors, imposes significant physical burdens on patients due to its locally invasive nature. Functional limitations are common, particularly in extremity and trunk locations, where tumors can restrict range of motion by up to 68% and cause muscle weakness, leading to difficulties in daily activities such as bathing, dressing, or carrying objects.[41][80] In cases involving the extremities, mobility loss is common, often resulting in reduced independence and the need for assistive devices. Post-surgical complications further exacerbate these issues, including infections and scarring. Chronic pain is a prevalent symptom, affecting up to 63% of patients and often described as severe and refractory to standard analgesics, contributing to sleep disturbances in 73% of those affected.[41][81] In intra-abdominal cases, which comprise 10-20% of all desmoid tumors, the infiltrative growth can lead to bowel or ureteral obstruction in 27-58% of patients, particularly those with familial adenomatous polyposis, potentially requiring urgent interventions like stenting or resection.[41][31] These physical manifestations not only limit mobility but also heighten fatigue and swelling around the tumor site, diminishing overall quality of life. The psychosocial impacts of aggressive fibromatosis are profound, with studies indicating heightened anxiety in 39% and depression in 50% of patients, significantly higher than in healthy controls.[82] Health-related quality of life (HRQoL) is impaired across multiple domains, including global health status (mean score 65.58 versus higher in controls), functional scales like physical and role functioning, and increased symptom burden from pain, fatigue, and insomnia.[83][41] Body image issues are particularly notable in abdominal wall cases, where visible lumps or surgical scars lead to feelings of disfigurement in 81% of patients and reduced femininity among women, affecting self-esteem and social interactions. Recurrence anxiety compounds these emotional challenges, stemming from the disease's unpredictable progression.[41] Economic consequences add to the burden, with high treatment costs—such as approximately $259,000 to prevent one disease progression with systemic therapies like sorafenib (based on number needed to treat)—and out-of-pocket expenses for surveillance imaging or travel to specialized centers.[55][41] Work absenteeism is substantial, as 26% of patients cease employment and 10% reduce to part-time due to pain and functional limitations, leading to financial strain and dependency on support systems.[84] Patient advocacy plays a crucial role in mitigating these impacts; the Desmoid Tumor Research Foundation provides education, peer support groups, and resources to empower patients and caregivers, fostering community and access to clinical trials.[85] Additionally, hormonal therapies, once more commonly used, raise fertility concerns, with 20% of affected women postponing pregnancy due to the disease and potential estrogen-modulating effects, necessitating individualized counseling.[41]Special Topics
Diagnostic coding
Aggressive fibromatosis, also known as desmoid tumor, is classified under neoplasms of uncertain behavior in medical coding systems to reflect its locally aggressive but non-metastasizing nature.[6] The primary ICD-10-CM code is D48.11 for desmoid tumor, encompassing aggressive fibromatosis across all sites.[86] Site-specific subcodes under D48.11 provide granularity based on anatomical location, facilitating precise clinical documentation; these include D48.110 for head and neck, D48.111 for chest wall, D48.112 for intrathoracic, D48.113 for abdominal wall, D48.114 for intra-abdominal, D48.115 for upper extremity and shoulder girdle, D48.116 for lower extremity and pelvic girdle, D48.117 for back, D48.118 for other site, and D48.119 for unspecified site.[87]| Code | Description |
|---|---|
| D48.110 | Desmoid tumor of head and neck |
| D48.111 | Desmoid tumor of chest wall |
| D48.112 | Desmoid tumor, intrathoracic |
| D48.113 | Desmoid tumor of abdominal wall |
| D48.114 | Desmoid tumor, intra-abdominal |
| D48.115 | Desmoid tumor of upper extremity and shoulder girdle |
| D48.116 | Desmoid tumor of lower extremity and pelvic girdle |
| D48.117 | Desmoid tumor of back |
| D48.118 | Desmoid tumor of other site |
| D48.119 | Desmoid tumor of unspecified site |