
Community hub
Recent from talks
Contribute something
Nothing was collected or created yet.
Phosphoinositide 3-kinase
View on Wikipedia| Phosphatidylinositol-4,5-bisphosphate 3-kinase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
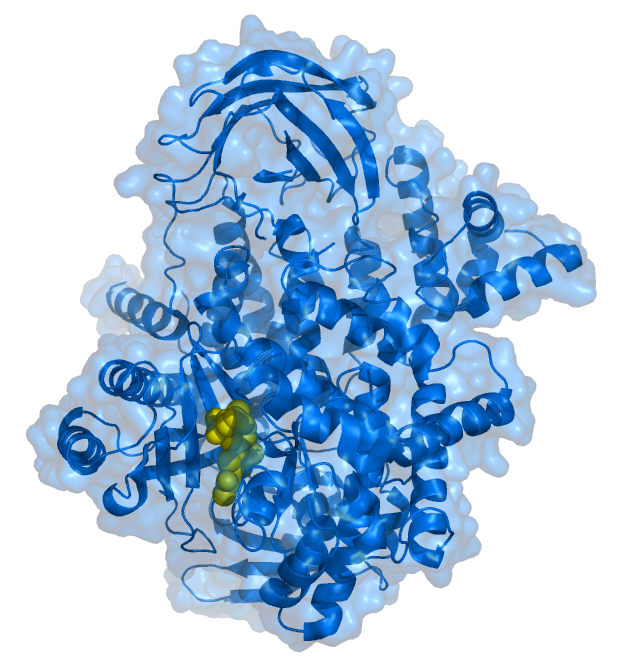
 PIK-93 inhibitor (yellow) bound to the PI3K 110 gamma subunit .[1] | |||||||||
| Identifiers | |||||||||
| Symbol | PI3K | ||||||||
| Pfam | PF00454 | ||||||||
| InterPro | IPR000403 | ||||||||
| SMART | SM00146 | ||||||||
| PROSITE | PDOC00710 | ||||||||
| SCOP2 | 3gmm / SCOPe / SUPFAM | ||||||||
| OPM superfamily | 265 | ||||||||
| OPM protein | 3ml9 | ||||||||
| |||||||||
| Phosphoinositide 3-kinase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| EC no. | 2.7.1.137 | ||||||||
| CAS no. | 115926-52-8 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| |||||||||
Phosphoinositide 3-kinases (PI3Ks), also called phosphatidylinositol 3-kinases, are a family of enzymes involved in cellular functions such as cell growth, proliferation, differentiation, motility, survival and intracellular trafficking, which in turn are involved in cancer.
PI3Ks are a family of related intracellular signal transducer enzymes capable of phosphorylating the 3 position hydroxyl group of the inositol ring of phosphatidylinositol (PtdIns).[2] The pathway, with oncogene PIK3CA and tumor suppressor gene PTEN, is implicated in the sensitivity of cancer tumors to insulin and IGF1, and in calorie restriction.[3][4]
Discovery
[edit]The discovery of PI3Ks by Lewis Cantley and colleagues began with their identification of a previously unknown phosphoinositide kinase associated with the polyoma middle T protein.[5] They observed unique substrate specificity and chromatographic properties of the products of the lipid kinase, leading to the discovery that this phosphoinositide kinase had the unprecedented ability to phosphorylate phosphoinositides on the 3' position of the inositol ring.[6] Subsequently, Cantley and colleagues demonstrated that in vivo the enzyme prefers PtdIns(4,5)P2 as a substrate, producing the novel phosphoinositide PtdIns(3,4,5)P3[7] previously identified in neutrophils.[8]
Classes
[edit]The PI3K family is divided into four different classes: Class I, Class II, Class III, and Class IV. The classifications are based on primary structure, regulation, and in vitro lipid substrate specificity.[9]
Class I
[edit]Class I PI3Ks catalyze the conversion of phosphatidylinositol (4,5)-bisphosphate (PI(4,5)P2) into phosphatidylinositol (3,4,5)-trisphosphate (PI(3,4,5)P3) in vivo. While in vitro, they have also been shown to convert phosphatidylinositol (PI) into phosphatidylinositol 3-phosphate (PI3P) and phosphatidylinositol 4-phosphate (PI4P) into phosphatidylinositol (3,4)-bisphosphate (PI(3,4)P2), these reactions are strongly disfavoured in vivo.[10][11][12][13] The PI3K is activated by G protein-coupled receptors and tyrosine kinase receptors.[9]
Class I PI3Ks are heterodimeric molecules composed of a regulatory and a catalytic subunit; they are further divided between IA and IB subsets on sequence similarity. Class IA PI3Ks are composed of a heterodimer between a p110 catalytic subunit and a shorter regulatory subunit (often p85).[14] There are five variants of the regulatory subunit: the three splice variants p85α, p55α, and p50α, p85β, and p55γ. There are also three variants of the p110 catalytic subunit designated p110α, β, or δ catalytic subunit. The first three regulatory subunits are all splice variants of the same gene (Pik3r1), the other two being expressed by other genes (Pik3r2 and Pik3r3, p85β, and p55γ, respectively). The most highly expressed regulatory subunit is p85α; all three catalytic subunits are expressed by separate genes (Pik3ca, Pik3cb, and Pik3cd for p110α, p110β, and p110δ, respectively). The first two p110 isoforms (α and β) are expressed in all cells, but p110δ is expressed primarily in leukocytes, and it has been suggested that it evolved in parallel with the adaptive immune system. The regulatory p101 and catalytic p110γ subunits comprise the class IB PI3Ks and are encoded by a single gene each (Pik3cg for p110γ and Pik3r5 for p101).
The p85 subunits contain SH2 and SH3 domains (Online Mendelian Inheritance in Man (OMIM): 171833). The SH2 domains bind preferentially to phosphorylated tyrosine residues in the amino acid sequence context Y-X-X-M.[15][16]
Classes II and III
[edit]Class II and III PI3Ks are differentiated from the Class I by their structure and function. The distinct feature of Class II PI3Ks is the C-terminal C2 domain. This domain lacks critical Asp residues to coordinate binding of Ca2+, which suggests class II PI3Ks bind lipids in a Ca2+-independent manner.
Class II comprises three catalytic isoforms (C2α, C2β, and C2γ), but, unlike Classes I and III, no regulatory proteins. Class II catalyse the production of PI(3)P from PI and PI(3,4)P2 from PI(4)P; however, little is known about their role in immune cells. PI(3,4)P2 has, however, been shown to play a role in the invagination phase of clathrin-mediated endocytosis.[17] C2α and C2β are expressed through the body, but expression of C2γ is limited to hepatocytes.
Class III PI3Ks produce only PI(3)P from PI [9] but are more similar to Class I in structure, as they exist as heterodimers of a catalytic (Vps34) and a regulatory (Vps15/p150) subunits. Class III seems to be primarily involved in the trafficking of proteins and vesicles. There is, however, evidence to show that they are able to contribute to the effectiveness of several process important to immune cells, not least phagocytosis.
Class IV
[edit]A group of more distantly related enzymes is sometimes referred to as class IV PI3Ks. It is composed of ataxia telangiectasia mutated (ATM), ataxia telangiectasia and Rad3 related (ATR), DNA-dependent protein kinase (DNA-PK) and mammalian target of rapamycin (mTOR). They are protein serine/threonine kinases.
Human genes
[edit]| group | gene | protein | aliases | EC number |
|---|---|---|---|---|
| class 1 catalytic | PIK3CA | PI3K, catalytic, alpha polypeptide | p110-α | 2.7.1.153 |
| PIK3CB | PI3K, catalytic, beta polypeptide | p110-β | ||
| PIK3CG | PI3K, catalytic, gamma polypeptide | p110-γ | ||
| PIK3CD | PI3K, catalytic, delta polypeptide | p110-δ | ||
| class 1 regulatory | PIK3R1 | PI3K, regulatory subunit 1 (alpha) | p85-α, p55-α, p50-α (splice variants) | N/A |
| PIK3R2 | PI3K, regulatory subunit 2 (beta) | p85-β | ||
| PIK3R3 | PI3K, regulatory subunit 3 (gamma) | p55-γ | ||
| PIK3R4 | PI3K, regulatory subunit 4 | p150 | ||
| PIK3R5 | PI3K, regulatory subunit 5 | p101 | ||
| PIK3R6 | PI3K, regulatory subunit 6 | p87 | ||
| class 2 | PIK3C2A | PI3K, class 2, alpha polypeptide | PI3K-C2α | 2.7.1.154 |
| PIK3C2B | PI3K, class 2, beta polypeptide | PI3K-C2β | ||
| PIK3C2G | PI3K, class 2, gamma polypeptide | PI3K-C2γ | ||
| class 3 | PIK3C3 | PI3K, class 3 | Vps34 | 2.7.1.137 |
Mechanism
[edit]The various 3-phosphorylated phosphoinositides that are produced by PI3Ks (PtdIns3P, PtdIns(3,4)P2, PtdIns(3,5)P2, and PtdIns(3,4,5)P3) function in a mechanism by which an assorted group of signalling proteins, containing PX domains, pleckstrin homology domains (PH domains), FYVE domains or other phosphoinositide-binding domains, are recruited to various cellular membranes.
Function
[edit]PI3Ks have been linked to an extraordinarily diverse group of cellular functions, including cell growth, proliferation, differentiation, motility, survival and intracellular trafficking. Many of these functions relate to the ability of class I PI3Ks to activate protein kinase B (PKB, aka Akt) as in the PI3K/AKT/mTOR pathway. The p110δ and p110γ isoforms regulate different aspects of immune responses. PI3Ks are also a key component of the insulin signaling pathway. Hence there is great interest in the role of PI3K signaling in diabetes mellitus. PI3K is also involved in interleukin signalling (IL4)[citation needed]
Mechanism
[edit]The pleckstrin homology domain of AKT binds directly to PtdIns(3,4,5)P3 and PtdIns(3,4)P2, which are produced by activated PI3Ks.[18] Since PtdIns(3,4,5)P3 and PtdIns(3,4)P2 are restricted to the plasma membrane, this results in translocation of AKT to the plasma membrane. Likewise, the phosphoinositide-dependent kinase-1 (PDK1 or, rarely referred to as PDPK1) also contains a pleckstrin homology domain that binds directly to PtdIns(3,4,5)P3 and PtdIns(3,4)P2, causing it to also translocate to the plasma membrane upon PI3K activation. The interaction of activated PDK1 and AKT allows AKT to become phosphorylated by PDK1 on threonine 308, leading to partial activation of AKT. Full activation of AKT occurs upon phosphorylation of serine 473 by the TORC2 complex of the mTOR protein kinase.
The PI3K/AKT pathway has been shown to be required for an extremely diverse array of cellular activities - most notably cellular proliferation and survival. For example, it was shown to be involved in the protection of astrocytes from ceramide-induced apoptosis.[19]
Many other proteins have been identified that are regulated by PtdIns(3,4,5)P3, including Bruton's tyrosine kinase (BTK), General Receptor for Phosphoinositides-1 (GRP1), and the O-linked N-acetylglucosamine (O-GlcNAc) transferase.
PtdIns(3,4,5)P3 also activates guanine‐nucleotide exchange factors (GEFs) that activate the GTPase Rac1,[20] leading to actin polymerization and cytoskeletal rearrangement.[21]
Cancers
[edit]The class IA PI3K p110α is mutated in many cancers. Many of these mutations cause the kinase to be more active. It is the single most mutated kinase in glioblastoma, the most malignant primary brain tumor.[22] The PtdIns(3,4,5)P3 phosphatase PTEN that antagonises PI3K signaling is absent from many tumours. In addition, the epidermal growth factor receptor EGFR that functions upstream of PI3K is mutationally activated or overexpressed in cancer.[22][23] Hence, PI3K activity contributes significantly to cellular transformation and the development of cancer. It has been shown that malignant B cells maintain a "tonic" activity of PI3K/Akt axis via upregulation of an adaptor protein GAB1, and this also allows B cells to survive targeted therapy with BCR inhibitors.[citation needed]
Learning and memory
[edit]PI3Ks have also been implicated in long-term potentiation (LTP). Whether they are required for the expression or the induction of LTP is still debated. In mouse hippocampal CA1 neurons, certain PI3Ks are complexed with AMPA receptors and compartmentalized at the postsynaptic density of glutamatergic synapses.[24] PI3Ks are phosphorylated upon NMDA receptor-dependent CaMKII activity,[25] and it then facilitates the insertion of AMPA-R GluR1 subunits into the plasma membrane. This suggests that PI3Ks are required for the expression of LTP. Furthermore, PI3K inhibitors abolished the expression of LTP in rat hippocampal CA1, but do not affect its induction.[26] Notably, the dependence of late-phase LTP expression on PI3Ks seems to decrease over time.[27]
However, another study found that PI3K inhibitors suppressed the induction, but not the expression, of LTP in mouse hippocampal CA1.[28] The PI3K pathway also recruits many other proteins downstream, including mTOR,[29] GSK3β,[30] and PSD-95.[29] The PI3K-mTOR pathway leads to the phosphorylation of p70S6K, a kinase that facilitates translational activity,[31][32] further suggesting that PI3Ks are required for the protein-synthesis phase of LTP induction instead.
PI3Ks interact with the insulin receptor substrate (IRS) to regulate glucose uptake through a series of phosphorylation events.
PI 3-kinases as protein kinases
[edit]Many PI3Ks appear to have a serine/threonine kinase activity in vitro; however, it is unclear whether this has any role in vivo.[citation needed]
Inhibition
[edit]All PI3Ks are inhibited by the drugs wortmannin and LY294002, although certain members of the class II PI3K family show decreased sensitivity. Wortmannin shows better efficiency than LY294002 on the hotspot mutation positions (GLU542, GLU545, and HIS1047)[33][34]
PI3K inhibitors as therapeutics
[edit]As wortmannin and LY294002 are broad-range inhibitors of PI3Ks and a number of unrelated proteins at higher concentrations, they are too toxic to be used as therapeutics.[citation needed] A number of pharmaceutical companies have thus developed PI3K isoform-specific inhibitors. As of January 2019, three PI3K inhibitors are approved by the FDA for routine clinical use in humans: the PIK3CD inhibitor idelalisib (July 2014, NDA 206545), the dual PIK3CA and PIK3CD inhibitor copanlisib (September 2017, NDA 209936), and the dual PIK3CD and PIK3CG inhibitor duvelisib (September 2018, NDA 211155). Co-targeted inhibition of the pathway with other pathways such as MAPK or PIM has been highlighted as a promising anti-cancer therapeutic strategy, which could offer benefit over the monotherapeutic approach by circumventing compensatory signalling, slowing the development of resistance and potentially allowing reduction of dosing.[35][36][37][38][39]
See also
[edit]References
[edit]- ^ PDB: 2chz; Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, et al. (May 2006). "A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling". Cell. 125 (4): 733–47. doi:10.1016/j.cell.2006.03.035. PMC 2946820. PMID 16647110.
- ^ "myo-inositol". Archived from the original on 2011-08-06. Retrieved 2006-01-28.
- ^ Giese N (2009). "Cell pathway on overdrive prevents cancer response to dietary restriction". PhysOrg.com. Retrieved 2009-04-22.
- ^ Kalaany NY, Sabatini DM (April 2009). "Tumours with PI3K activation are resistant to dietary restriction". Nature. 458 (7239): 725–31. Bibcode:2009Natur.458..725K. doi:10.1038/nature07782. PMC 2692085. PMID 19279572.
- ^ Whitman M, Kaplan DR, Schaffhausen B, Cantley L, Roberts TM (1985). "Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation". Nature. 315 (6016): 239–42. Bibcode:1985Natur.315..239W. doi:10.1038/315239a0. PMID 2987699. S2CID 4337999.
- ^ Whitman M, Downes CP, Keeler M, Keller T, Cantley L (April 1988). "Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate". Nature. 332 (6165): 644–6. Bibcode:1988Natur.332..644W. doi:10.1038/332644a0. PMID 2833705. S2CID 4326568.
- ^ Auger KR, Serunian LA, Soltoff SP, Libby P, Cantley LC (April 1989). "PDGF-dependent tyrosine phosphorylation stimulates production of novel polyphosphoinositides in intact cells". Cell. 57 (1): 167–75. doi:10.1016/0092-8674(89)90182-7. PMID 2467744. S2CID 22154860.
- ^ Traynor-Kaplan AE, Harris AL, Thompson BL, Taylor P, Sklar LA (July 1988). "An inositol tetrakisphosphate-containing phospholipid in activated neutrophils". Nature. 334 (6180): 353–6. Bibcode:1988Natur.334..353T. doi:10.1038/334353a0. PMID 3393226. S2CID 4263472.
- ^ a b c Leevers SJ, Vanhaesebroeck B, Waterfield MD (April 1999). "Signalling through phosphoinositide 3-kinases: the lipids take centre stage". Current Opinion in Cell Biology. 11 (2): 219–25. doi:10.1016/S0955-0674(99)80029-5. PMID 10209156.
- ^ Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT (August 2017). "The PI3K Pathway in Human Disease". Cell. 170 (4): 605–635. doi:10.1016/j.cell.2017.07.029. PMC 5726441. PMID 28802037.
- ^ Jean S, Kiger AA (March 2014). "Classes of phosphoinositide 3-kinases at a glance". Journal of Cell Science. 127 (Pt 5): 923–8. doi:10.1242/jcs.093773. PMC 3937771. PMID 24587488.
- ^ Vanhaesebroeck B, Stephens L, Hawkins P (February 2012). "PI3K signalling: the path to discovery and understanding". Nature Reviews. Molecular Cell Biology. 13 (3): 195–203. doi:10.1038/nrm3290. PMID 22358332. S2CID 6999833.
- ^ Okkenhaug K (January 2013). "Signaling by the phosphoinositide 3-kinase family in immune cells". Annual Review of Immunology. 31 (2): 675–704. doi:10.1146/annurev-immunol-032712-095946. PMC 4516760. PMID 23330955.
- ^ Carpenter CL, Duckworth BC, Auger KR, Cohen B, Schaffhausen BS, Cantley LC (November 1990). "Purification and characterization of phosphoinositide 3-kinase from rat liver". The Journal of Biological Chemistry. 265 (32): 19704–11. doi:10.1016/S0021-9258(17)45429-9. PMID 2174051.
- ^ Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, et al. (March 1993). "SH2 domains recognize specific phosphopeptide sequences". Cell. 72 (5): 767–78. doi:10.1016/0092-8674(93)90404-E. PMID 7680959.
- ^ Yoakim M, Hou W, Songyang Z, Liu Y, Cantley L, Schaffhausen B (September 1994). "Genetic analysis of a phosphatidylinositol 3-kinase SH2 domain reveals determinants of specificity". Molecular and Cellular Biology. 14 (9): 5929–38. doi:10.1128/MCB.14.9.5929. PMC 359119. PMID 8065326.
- ^ Posor Y, Eichhorn-Grünig M, Haucke V (June 2015). "Phosphoinositides in endocytosis". Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 1851 (6): 794–804. doi:10.1016/j.bbalip.2014.09.014. PMID 25264171.
- ^ Franke TF, Kaplan DR, Cantley LC, Toker A (January 1997). "Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate". Science. 275 (5300): 665–8. doi:10.1126/science.275.5300.665. PMID 9005852. S2CID 31186873.
- ^ Gómez Del Pulgar T, De Ceballos ML, Guzmán M, Velasco G (September 2002). "Cannabinoids protect astrocytes from ceramide-induced apoptosis through the phosphatidylinositol 3-kinase/protein kinase B pathway". The Journal of Biological Chemistry. 277 (39): 36527–33. doi:10.1074/jbc.M205797200. hdl:10261/36487. PMID 12133838.
- ^ Welch HC, Coadwell WJ, Stephens LR, Hawkins PT (July 2003). "Phosphoinositide 3-kinase-dependent activation of Rac". FEBS Letters. 546 (1): 93–7. Bibcode:2003FEBSL.546...93W. doi:10.1016/s0014-5793(03)00454-x. PMID 12829242.
- ^ Jaffe AB, Hall A (2005). "Rho GTPases: biochemistry and biology". Annual Review of Cell and Developmental Biology. 21: 247–69. doi:10.1146/annurev.cellbio.21.020604.150721. PMID 16212495.
- ^ a b Bleeker FE, Lamba S, Zanon C, Molenaar RJ, Hulsebos TJ, Troost D, et al. (September 2014). "Mutational profiling of kinases in glioblastoma". BMC Cancer. 14 718. doi:10.1186/1471-2407-14-718. PMC 4192443. PMID 25256166.
- ^ Bleeker FE, Molenaar RJ, Leenstra S (May 2012). "Recent advances in the molecular understanding of glioblastoma". Journal of Neuro-Oncology. 108 (1): 11–27. doi:10.1007/s11060-011-0793-0. PMC 3337398. PMID 22270850.
- ^ Man HY, Wang Q, Lu WY, Ju W, Ahmadian G, Liu L, et al. (May 2003). "Activation of PI3-kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons". Neuron. 38 (4): 611–24. doi:10.1016/S0896-6273(03)00228-9. PMID 12765612.
- ^ Joyal JL, Burks DJ, Pons S, Matter WF, Vlahos CJ, White MF, Sacks DB (November 1997). "Calmodulin activates phosphatidylinositol 3-kinase". The Journal of Biological Chemistry. 272 (45): 28183–6. doi:10.1074/jbc.272.45.28183. PMID 9353264.
- ^ Sanna PP, Cammalleri M, Berton F, Simpson C, Lutjens R, Bloom FE, Francesconi W (May 2002). "Phosphatidylinositol 3-kinase is required for the expression but not for the induction or the maintenance of long-term potentiation in the hippocampal CA1 region". The Journal of Neuroscience. 22 (9): 3359–65. doi:10.1523/JNEUROSCI.22-09-03359.2002. PMC 6758361. PMID 11978812.
- ^ Karpova A, Sanna PP, Behnisch T (February 2006). "Involvement of multiple phosphatidylinositol 3-kinase-dependent pathways in the persistence of late-phase long term potentiation expression". Neuroscience. 137 (3): 833–41. doi:10.1016/j.neuroscience.2005.10.012. PMID 16326012. S2CID 38232127.
- ^ Opazo P, Watabe AM, Grant SG, O'Dell TJ (May 2003). "Phosphatidylinositol 3-kinase regulates the induction of long-term potentiation through extracellular signal-related kinase-independent mechanisms". The Journal of Neuroscience. 23 (9): 3679–88. doi:10.1523/JNEUROSCI.23-09-03679.2003. PMC 6742185. PMID 12736339.
- ^ a b Yang PC, Yang CH, Huang CC, Hsu KS (February 2008). "Phosphatidylinositol 3-kinase activation is required for stress protocol-induced modification of hippocampal synaptic plasticity". The Journal of Biological Chemistry. 283 (5): 2631–43. doi:10.1074/jbc.M706954200. PMID 18057005.
- ^ Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, et al. (March 2007). "LTP inhibits LTD in the hippocampus via regulation of GSK3beta". Neuron. 53 (5): 703–17. doi:10.1016/j.neuron.2007.01.029. PMID 17329210. S2CID 6903401.
- ^ Toker A, Cantley LC (June 1997). "Signalling through the lipid products of phosphoinositide-3-OH kinase". Nature. 387 (6634): 673–6. Bibcode:1997Natur.387..673T. doi:10.1038/42648. PMID 9192891. S2CID 4347728.
- ^ Cammalleri M, Lütjens R, Berton F, King AR, Simpson C, Francesconi W, Sanna PP (November 2003). "Time-restricted role for dendritic activation of the mTOR-p70S6K pathway in the induction of late-phase long-term potentiation in the CA1". Proceedings of the National Academy of Sciences of the United States of America. 100 (24): 14368–73. Bibcode:2003PNAS..10014368C. doi:10.1073/pnas.2336098100. PMC 283598. PMID 14623952.
- ^ Kumar DT, Doss CG (2016-01-01). Investigating the Inhibitory Effect of Wortmannin in the Hotspot Mutation at Codon 1047 of PIK3CA Kinase Domain: A Molecular Docking and Molecular Dynamics Approach. Vol. 102. pp. 267–97. doi:10.1016/bs.apcsb.2015.09.008. ISBN 9780128047958. PMID 26827608.
{{cite book}}:|journal=ignored (help) - ^ Sudhakar N, Priya Doss CG, Thirumal Kumar D, Chakraborty C, Anand K, Suresh M (2016-01-02). "Deciphering the impact of somatic mutations in exon 20 and exon 9 of PIK3CA gene in breast tumors among Indian women through molecular dynamics approach". Journal of Biomolecular Structure & Dynamics. 34 (1): 29–41. doi:10.1080/07391102.2015.1007483. PMID 25679319. S2CID 205575161.
- ^ Malone T, Schäfer L, Simon N, Heavey S, Cuffe S, Finn S, et al. (March 2020). "Current perspectives on targeting PIM kinases to overcome mechanisms of drug resistance and immune evasion in cancer" (PDF). Pharmacology & Therapeutics. 207 107454. doi:10.1016/j.pharmthera.2019.107454. PMID 31836451. S2CID 209357486.
- ^ Luszczak S, Kumar C, Sathyadevan VK, Simpson BS, Gately KA, Whitaker HC, Heavey S (2020). "PIM kinase inhibition: co-targeted therapeutic approaches in prostate cancer". Signal Transduction and Targeted Therapy. 5 7. doi:10.1038/s41392-020-0109-y. PMC 6992635. PMID 32025342.
- ^ Heavey S, Dowling P, Moore G, Barr MP, Kelly N, Maher SG, et al. (January 2018). "Development and characterisation of a panel of phosphatidylinositide 3-kinase - mammalian target of rapamycin inhibitor resistant lung cancer cell lines". Scientific Reports. 8 (1) 1652. Bibcode:2018NatSR...8.1652H. doi:10.1038/s41598-018-19688-1. PMC 5786033. PMID 29374181.
- ^ Heavey S, Godwin P, Baird AM, Barr MP, Umezawa K, Cuffe S, et al. (October 2014). "Strategic targeting of the PI3K-NFκB axis in cisplatin-resistant NSCLC". Cancer Biology & Therapy. 15 (10): 1367–77. doi:10.4161/cbt.29841. PMC 4130730. PMID 25025901.
- ^ Heavey S, O'Byrne KJ, Gately K (April 2014). "Strategies for co-targeting the PI3K/AKT/mTOR pathway in NSCLC". Cancer Treatment Reviews. 40 (3): 445–56. doi:10.1016/j.ctrv.2013.08.006. PMID 24055012.
Further reading
[edit]- Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, et al. (2001). "Synthesis and function of 3-phosphorylated inositol lipids". Annual Review of Biochemistry. 70: 535–602. doi:10.1146/annurev.biochem.70.1.535. PMID 11395417. [1]
- Schild C, Wirth M, Reichert M, Schmid RM, Saur D, Schneider G (December 2009). "PI3K signaling maintains c-myc expression to regulate transcription of E2F1 in pancreatic cancer cells". Molecular Carcinogenesis. 48 (12): 1149–58. doi:10.1002/mc.20569. PMID 19603422. S2CID 41545085.
- Williams R, Berndt A, Miller S, Hon WC, Zhang X (August 2009). "Form and flexibility in phosphoinositide 3-kinases". Biochemical Society Transactions. 37 (Pt 4): 615–26. doi:10.1042/BST0370615. PMID 19614567.
- Quaresma AJ, Sievert R, Nickerson JA (April 2013). "Regulation of mRNA export by the PI3 kinase/AKT signal transduction pathway". Molecular Biology of the Cell. 24 (8): 1208–21. doi:10.1091/mbc.E12-06-0450. PMC 3623641. PMID 23427269.
External links
[edit]- Eukaryotic Linear Motif resource motif class MOD_PIKK_1
- Proteopedia Phosphoinositide_3-Kinases to explore the structure in interactive 3D
- PI-3+Kinase at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- PI3K/Akt Signaling Pathway
Phosphoinositide 3-kinase
View on GrokipediaHistory and Discovery
Initial Discovery
The discovery of phosphoinositide 3-kinase (PI3K) activity took place in 1988, during investigations into the mechanisms of cellular transformation by polyomavirus middle T antigen. Researchers in Lewis Cantley's laboratory at Tufts University, led by Malcolm Whitman and colleagues, identified a novel lipid kinase activity tightly associated with the middle T antigen in immunoprecipitates from transformed cells. This enzyme phosphorylated the D-3 position of the inositol ring in phosphatidylinositol (PI), generating the previously unknown lipid phosphatidylinositol 3-phosphate (PI(3)P).[4] Biochemical assays employed in these studies utilized radiolabeled ATP and thin-layer chromatography to detect the phosphorylation products, confirming the reaction's dependence on ATP and specificity for the 3-position, distinguishing it from known type I and type II PI kinases that target the 4- or 5-positions. This finding supported the emerging lipid second messenger hypothesis championed by Cantley's group, proposing that 3-phosphorylated phosphoinositides act as signaling molecules in processes like viral transformation and receptor tyrosine kinase pathways.[4] By 1991, significant progress was made in purifying and cloning the enzyme from bovine brain tissue. J. Alberto Escobedo and colleagues at Genentech isolated cDNA encoding the 85 kDa regulatory subunit (p85), revealing PI3K as a heterodimeric complex comprising this p85 subunit and an associated 110 kDa catalytic subunit (p110). The p85 subunit featured SH2-like motifs enabling binding to phosphotyrosine residues on activated receptors, such as the platelet-derived growth factor receptor.[5] Further early biochemical characterization, using purified enzyme preparations, demonstrated PI3K's substrate versatility: it catalyzes ATP-dependent phosphorylation of PI to yield PI(3)P, PI(4)P to PI(3,4)P2, and PI(4,5)P2 to PI(3,4,5)P3. These reactions were quantified via in vitro kinase assays with lipid vesicles and HPLC analysis of deacylated products, establishing the enzyme's role in generating multiple 3-phosphoinositide signals.[6]Classification Development
The classification of phosphoinositide 3-kinases (PI3Ks) into distinct classes began in the mid-1990s, as cloning efforts revealed a diverse family of enzymes beyond the initial p110 subunit identified in signaling complexes. Early work distinguished PI3Ks based on shared catalytic domains but varying accessory regions, regulatory interactions, and substrate specificities, such as preferences for phosphatidylinositol (PI), PI(4)P, or PI(4,5)P2. A seminal 1999 review by Rameh and Cantley formalized this class-based approach, grouping the enzymes into categories that reflected their structural motifs—like Ras-binding domains in class I—and modes of activation, such as receptor tyrosine kinase coupling versus G-protein regulation. This framework shifted focus from isolated activities to a unified family model, enabling comparative analyses across isoforms.[7] Refinements to the classification accelerated in the 2000s, driven by genomic sequencing projects that identified PI3K orthologs in diverse eukaryotes, from yeast to mammals, and highlighted evolutionary divergences. Vanhaesebroeck et al. in 2001 consolidated the evidence into a four-class system (I–IV), where class I enzymes form heterodimers activated by growth factors, class II features C-terminal extensions for membrane localization, class III specializes in PI(3)P production for trafficking, and class IV encompasses atypical kinases with mixed lipid/protein activities. This 2001 synthesis integrated phylogenetic data and functional assays, establishing the classes as structurally and biochemically discrete. Subsequent updates, including a 2005 analysis by the same group, incorporated knockout mouse studies to validate class distinctions through isoform-specific phenotypes in immunity and metabolism.[8][9] Nomenclature evolved alongside these milestones to reflect subunit compositions and conservation. For instance, class I catalytic subunits are designated p110α (encoded by PIK3CA), p110β (PIK3CB), p110δ (PIK3CD), and p110γ (PIK3CG), paired with regulatory partners like p85 or p101; class III is uniformly VPS34 (PIK3C3), often complexed with VPS15. The Human Genome Project's completion in 2003 enabled comprehensive isoform mapping, confirming eight human catalytic isoforms and resolving ambiguities in orthology, such as the absence of class I in yeast. These conventions, codified in the 2001 review, persist in current literature for precision in genetic and pharmacological studies. Evolutionary insights further shaped classification by tracing the family's origins to ancient eukaryotic processes. Class III PI3K, exemplified by yeast Vps34 identified in 1993, demonstrates remarkable conservation, with its mammalian ortholog retaining roles in endosomal sorting and autophagy across 1.5 billion years of divergence. This deep homology contrasted with the expansion of classes I and II in metazoans, linked to multicellular signaling needs, reinforcing the 2001 four-class paradigm as evolutionarily grounded.[10]Structure and Classification
Class I PI3Ks
Class I PI3Ks are the most extensively studied subclass of phosphoinositide 3-kinases, functioning primarily as signaling enzymes that generate second messengers in response to extracellular stimuli. They operate as obligate heterodimers composed of a catalytic p110 subunit and a regulatory subunit, which together confer specificity in activation and localization. This subclass is divided into two main groups: class IA and class IB. Class IA PI3Ks include the catalytic isoforms p110α (encoded by PIK3CA), p110β (PIK3CB), and p110δ (PIK3CD), which pair with regulatory subunits from the p85 family, such as p85α (and its splice variants p55α and p50α), p85β, or p55γ. In contrast, class IB PI3Ks consist of the catalytic isoform p110γ (PIK3CG) associated with regulatory subunits p101 or p84 (also known as p87).[1] The structural architecture of class I PI3Ks is characterized by distinct domains that enable regulation and substrate interaction. The p110 catalytic subunit features an N-terminal regulatory region with a p85-binding domain (for class IA) or p101/p84-binding domain (for class IB), followed by a Ras-binding domain (RBD) that mediates interaction with GTP-bound Ras proteins, a C2 domain for membrane association, a helical domain, and a C-terminal lipid kinase domain responsible for phosphotransfer. The regulatory subunits play a crucial inhibitory role in the basal state by binding to the p110 helical domain, suppressing activity; upon activation, this inhibition is relieved. In class IA, the p85 regulatory subunits contain two Src homology 2 (SH2) domains (N-terminal and C-terminal) flanking an inter-SH2 (iSH2) region that interacts with p110, allowing recruitment to phosphotyrosine motifs. Class IB regulatory subunits, p101 and p84, lack SH2 domains but possess helical regions that stabilize the complex and facilitate G-protein interactions. Recent cryo-EM structures have revealed conformational changes in PI3Kα upon RAS binding, highlighting isoform-specific interactions that enhance activation and providing insights for targeted inhibition.[1][11][12] Class I PI3Ks exhibit high substrate specificity, primarily phosphorylating phosphatidylinositol 4,5-bisphosphate [PI(4,5)P₂] at the 3-position of the inositol ring to produce phosphatidylinositol 3,4,5-trisphosphate [PI(3,4,5)P₃], a key lipid second messenger that recruits downstream effectors like Akt via pleckstrin homology domains. They also generate PI(3,4)P₂ indirectly through dephosphorylation of PI(3,4,5)P₃. Activation mechanisms differ between subclasses: class IA PI3Ks are primarily recruited and activated by receptor tyrosine kinases (RTKs), where phosphotyrosine residues on the receptor or adaptor proteins bind the SH2 domains of p85, leading to allosteric relief of inhibition and membrane localization. Class IB PI3Ks, particularly p110γ, are activated downstream of G-protein-coupled receptors (GPCRs) through direct binding of Gβγ subunits to the p110γ catalytic subunit or via the p101/p84 regulators, enabling rapid signaling in response to chemoattractants. Additionally, all class I isoforms can be modulated by Ras binding to the RBD, enhancing activity.[1][13] Tissue distribution of class I PI3K isoforms reflects their specialized roles in cellular signaling. The p110α and p110β isoforms are ubiquitously expressed across mammalian tissues, supporting broad housekeeping and growth-related functions. In contrast, p110δ is predominantly enriched in cells of the hematopoietic lineage, particularly B lymphocytes, where it contributes to antigen receptor signaling. The p110γ isoform is highly expressed in myeloid cells, such as neutrophils and macrophages, aligning with its role in immune responses to inflammatory signals.[1][14][11]Class II PI3Ks
Class II phosphoinositide 3-kinases (PI3Ks) are a distinct subfamily of enzymes characterized by their monomeric structure and specialized roles in cellular membrane dynamics. In humans, this class includes three isoforms: PI3K-C2α (encoded by PIK3C2A), PI3K-C2β (encoded by PIK3C2B), and PI3K-C2γ (encoded by PIK3C2G). Unlike class I PI3Ks, which form heterodimers with regulatory subunits, class II PI3Ks consist of single polypeptide chains lacking p85-like regulators, enabling autonomous activity modulated primarily through intramolecular interactions and membrane association.[15][16] Structurally, class II PI3Ks feature a conserved catalytic core comprising a helical domain and a kinase domain, flanked by lipid-binding motifs that facilitate their recruitment to cellular membranes. All isoforms possess at least one C2 domain, which mediates calcium-dependent binding to phospholipid membranes, while PI3K-C2α and PI3K-C2β additionally contain a Phox homology (PX) domain that interacts with specific phosphoinositides to enhance localization. These domains distinguish class II PI3Ks from other classes by promoting direct engagement with endomembranes, such as clathrin-coated vesicles in the case of PI3K-C2α. The absence of regulatory subunits contrasts with the heterodimeric architecture of class I enzymes, underscoring their adaptation for localized, trafficking-oriented functions rather than broad signaling cascades.[15][17][16] Functionally, class II PI3Ks exhibit substrate preferences that support their involvement in membrane trafficking, phosphorylating phosphatidylinositol (PI) to generate PI(3)P and PI(4)P to produce PI(3,4)P2. These products recruit effector proteins to endocytic compartments, facilitating vesicle formation and maturation. Notably, class II enzymes display reduced sensitivity to the inhibitor wortmannin at low concentrations (typically <100 nM), allowing selective inhibition studies that highlight their distinct pharmacology compared to more sensitive class I and III PI3Ks.[15][18][16] Regulation of class II PI3Ks is tightly linked to endocytic processes, with PI3K-C2α and PI3K-C2β associating with clathrin via an N-terminal clathrin-binding domain, positioning them at sites of vesicle budding. Activation occurs through upstream signals from integrins and growth factors, such as epidermal growth factor (EGF) and insulin, which promote their recruitment to early endosomes and recycling pathways via receptor tyrosine kinase interactions. This clathrin-dependent mechanism ensures localized PI(3,4)P2 production, which stabilizes membrane curvature during trafficking events without the receptor-mediated dimerization seen in class I PI3Ks.[15][19][16]Class III PI3Ks
Class III PI3Ks consist of a single catalytic isoform, known as VPS34 or hVps34 in humans, encoded by the PIK3C3 gene.[20] This enzyme forms multi-subunit complexes essential for its localization and activity, primarily including the regulatory subunit VPS15 (also called p150 or PIK3R4), which stabilizes VPS34 and facilitates membrane association.[20] Additional core components include Beclin-1 (encoded by BECN1), which acts as a scaffold for complex assembly, and UVRAG (UV radiation resistance-associated gene), which modulates specificity within different complexes.[20] These interactions enable VPS34 to participate in distinct assemblies: one involving Beclin-1 and ATG14L for autophagy initiation, and another with Beclin-1 and UVRAG for endocytic regulation.[20] Structurally, VPS34 features a conserved architecture with a C2 domain for membrane binding, a helical domain, and a kinase domain comprising N- and C-lobes with an ATP-binding pocket. The helical and kinase domains exhibit similarity to those in class I PI3Ks, including an ordered activation loop that positions the catalytic residues, but VPS34 lacks the Ras-binding domain present in class I isoforms, reflecting its independence from Ras-mediated signaling.[20] Its activity is tightly regulated by nutrient-sensing pathways, particularly through inhibition by mTORC1 under nutrient-replete conditions, which phosphorylates accessory subunits like ATG14L and NRBF2 to suppress VPS34 function.[20] VPS34 exclusively phosphorylates phosphatidylinositol (PI) at the 3-position of the inositol ring to generate phosphatidylinositol 3-phosphate [PI(3)P], its sole lipid substrate.[20] This product is crucial for recruiting effector proteins that drive autophagosome formation and endosome maturation, thereby coordinating vesicular trafficking events.[20] The enzyme's role in these processes underscores its evolutionary conservation, with orthologs tracing back to the yeast Vps34 protein, first identified for its defects in vacuolar protein sorting.[21] This ancient function highlights VPS34's fundamental involvement in eukaryotic membrane dynamics, preserved from yeast to humans across diverse orthologous complexes.[20]PI3K-related kinases (sometimes referred to as Class IV)
PI3K-related kinases (PIKKs), sometimes referred to as atypical or Class IV in certain contexts due to sequence homology in their catalytic domains, encompass a group of enzymes that diverge significantly from the canonical PI3K classes (I-III) in both structure and function. These are primarily serine/threonine protein kinases rather than lipid kinases. The main members include the mammalian target of rapamycin (mTOR), ataxia-telangiectasia mutated (ATM), ATM- and Rad3-related (ATR), DNA-dependent protein kinase catalytic subunit (DNA-PKcs), suppressor with morphogenetic effect on genitalia 1 (SMG1), and transformation/transcription domain-associated protein (TRRAP). These kinases share limited conservation of the classic PI3K kinase domain, with homology primarily in the catalytic core, which justifies their grouping as related to PI3Ks despite their shift toward protein phosphorylation. Phosphatidylinositol 4-kinases (PI4Ks) are related lipid kinases but classified separately, as they focus on 4-phosphorylation of inositol.[22][23][24] Structurally, PIKKs are large proteins (280–470 kDa) featuring a conserved C-terminal region with FAT (FRAP-ATM-TRRAP), PI3K-like kinase, and FATC domains, alongside an N-terminal array of HEAT repeats that mediate protein-protein interactions. For example, mTOR includes extensive HEAT repeats and FAT domains that enable binding to rapamycin-FKBP12 complexes, facilitating regulation within mTORC1 and mTORC2 assemblies for nutrient sensing and growth control. DNA-PKcs, in contrast, possesses a prominent kinase domain integrated into a circular cradle structure flanked by FAT and FATC domains, allowing it to associate with the Ku70/Ku80 heterodimer for DNA end recognition during repair. This architectural divergence supports their specialized roles, such as mTOR's integration into multi-subunit complexes versus the DNA-binding capabilities of ATM, ATR, and DNA-PKcs.[22][25][26] In terms of activity, PIKKs exhibit minimal lipid kinase function and instead primarily phosphorylate protein substrates to regulate cellular processes. mTOR, for instance, directly phosphorylates ribosomal protein S6 kinase (S6K) and eukaryotic initiation factor 4E-binding protein 1 (4E-BP1), promoting cap-dependent translation initiation and protein synthesis essential for cell growth. ATM, ATR, and DNA-PKcs activate the DNA damage response by phosphorylating targets like p53, CHK1, and CHK2, coordinating cell cycle arrest, apoptosis, and non-homologous end joining repair. SMG1 contributes to nonsense-mediated mRNA decay, while TRRAP acts in a catalytically inactive manner to support histone acetyltransferase complexes for transcription.[22][27] The rationale for their relation to PI3Ks lies in the sequence homology of their catalytic cores to canonical PI3Ks, enabling shared sensitivity to inhibitors like wortmannin (with higher IC50 values compared to classes I-III), while their functions have evolved toward protein-centric regulation of DNA damage response, growth, and mRNA surveillance. This homology underscores their evolutionary relation to lipid kinases but highlights a functional adaptation for intracellular signaling in stress and proliferation contexts.[22][23]Genetics and Molecular Architecture
Human Encoding Genes
In humans, the phosphoinositide 3-kinases (PI3Ks) are encoded by a set of genes that produce catalytic and regulatory subunits, with distinct loci across the genome corresponding to the four main classes. These genes are essential for assembling the heterodimeric or monomeric complexes characteristic of each class. The catalytic subunits are primarily encoded by genes in the PIK3C family, while regulatory subunits are encoded by PIK3R genes for class I. Pseudogenes, such as a processed pseudogene of PIK3CA on chromosome 22, also exist but do not produce functional proteins.[28] The following table summarizes the primary human encoding genes for PI3K subunits, including their chromosomal locations:| Class | Subunit Type | Gene Symbol | Protein Name | Chromosomal Location |
|---|---|---|---|---|
| I (IA) | Catalytic | PIK3CA | p110α | 3q26.32[29] |
| I (IA) | Catalytic | PIK3CB | p110β | 3q22.3[30] |
| I (IA) | Catalytic | PIK3CD | p110δ | 1p36.22[31] |
| I (IB) | Catalytic | PIK3CG | p110γ | 7q22.3[32] |
| I (IA) | Regulatory | PIK3R1 | p85α, p55α, p50α | 5q13.1[33] |
| I (IA) | Regulatory | PIK3R2 | p85β | 19p13.11[34] |
| I (IA) | Regulatory | PIK3R3 | p55γ | 1p34.1[35] |
| I (IB) | Regulatory | PIK3R5 | p101 | 17p13.1[36] |
| I (IB) | Regulatory | PIK3R6 | p84 | 17p13.1[37] |
| II | Catalytic | PIK3C2A | PI3K-C2α | 11p15.1[38] |
| II | Catalytic | PIK3C2B | PI3K-C2β | 1q32.1[39] |
| II | Catalytic | PIK3C2G | PI3K-C2γ | 12p12.3[40] |
| III | Catalytic | PIK3C3 | VPS34 | 18q12.3[41] |
Domain Structure and Isoforms
The catalytic subunits of phosphoinositide 3-kinases (PI3Ks) share a conserved modular architecture that facilitates their recruitment to cellular membranes and execution of lipid phosphorylation. Central to this structure is the Ras-binding domain (RBD), which interacts with GTP-bound Ras proteins to enhance membrane localization and activation, particularly in class I and II isoforms.[48] Adjacent to the RBD is the C2 domain, a phospholipid-binding module that promotes membrane targeting through interactions with negatively charged lipids like phosphatidylserine, thereby positioning the enzyme for substrate access.[1] The helical domain follows, serving as a structural scaffold that interfaces with regulatory elements, while the C-terminal kinase domain harbors the catalytic core responsible for phosphorylating the 3-position of the inositol ring in phosphoinositides.[49] These domains form the PI3K signature motif, a hallmark conserved across classes I, II, and III, though class III enzymes like VPS34 lack the RBD and C2 domains, relying instead on a simpler helical-kinase arrangement within multi-subunit complexes.[1] In class IA PI3Ks, regulatory interactions are mediated by the p85 family of adaptor subunits, which contain two Src homology 2 (SH2) domains that bind phosphotyrosine (pTyr) motifs on activated receptor tyrosine kinases or adaptor proteins such as IRS1, thereby recruiting the p110 catalytic subunit to signaling hotspots at the plasma membrane.[50] These SH2-pTyr interactions relieve autoinhibitory interfaces, particularly between the nSH2 domain of p85 and the helical/kinase domains of p110, which in the basal state sterically hinder ATP and substrate binding in the catalytic pocket.[51] Activation disrupts this inhibition, allowing conformational rearrangements that expose the active site, as evidenced by structural analyses showing nSH2 displacement upon pTyr engagement.[49] Such regulatory mechanisms ensure tight control over PI3K activity, preventing aberrant signaling. Isoform diversity arises from distinct catalytic subunits and splice variants that modulate domain functionality and tissue specificity. Class I PI3Ks include p110α (encoded by PIK3CA), p110β (PIK3CB), p110δ (PIK3CD), and the class IB p110γ (PIK3CG), each sharing the core RBD-C2-helical-kinase architecture but differing in regulatory subunit pairing and Ras affinity—p110α and p110γ exhibit strong RBD-Ras interactions, while p110β and p110δ show weaker binding.[52] Splice variants further diversify function; for instance, alternative splicing of PIK3CA produces truncated p110α isoforms lacking portions of the C-terminal regulatory region, altering stability and autoinhibitory interactions compared to the full-length 1068-amino-acid form.[53] In class II PI3Ks (PI3K-C2α, -C2β, -C2γ), an extended N-terminal region precedes the RBD, and the C2 domain uniquely incorporates lipid-sensing elements, with evidence suggesting calcium (Ca²⁺)-dependent conformational changes in PI3K-C2α that enhance membrane association under elevated intracellular Ca²⁺ levels.[54] Structural studies, particularly using cryo-electron microscopy (cryo-EM), have illuminated these domain arrangements in near-native states. For example, the cryo-EM structure of the p110γ-p101 complex (PDB ID: 7MEZ) at 3.7 Å resolution depicts the compact assembly of RBD, C2, helical, and kinase domains, highlighting the PIP₂-binding pocket within the kinase domain formed by conserved residues in the activation loop.[55] Similar models for class I p110α reveal how helical domain mutations disrupt autoinhibitory contacts, promoting constitutive activity observed in oncogenic contexts.[51] These insights underscore the architectural basis for isoform-specific regulation and therapeutic targeting.Catalytic Mechanisms
Lipid Kinase Activity
Phosphoinositide 3-kinases (PI3Ks) exert their primary function as lipid kinases by transferring the γ-phosphate group from ATP to the D-3 hydroxyl position of the inositol ring in phosphoinositide substrates. This phosphorylation generates key second messengers that regulate diverse cellular processes. A prototypical reaction catalyzed by class I PI3Ks is the conversion of phosphatidylinositol 4,5-bisphosphate (PI(4,5)P₂) to phosphatidylinositol 3,4,5-trisphosphate (PI(3,4,5)P₃): This reaction was first characterized in activated neutrophils, where PI(3,4,5)P₃ accumulation was shown to arise directly from PI(4,5)P₂ phosphorylation.[56] Substrate specificity differs across PI3K classes, reflecting their distinct physiological roles. Class I PI3Ks exhibit high preference for PI(4,5)P₂, with Michaelis constant (Kₘ) values typically ranging from 1 to 10 μM, as measured in recombinant p110α assays using sonicated lipid vesicles and radiolabeled ATP. For instance, purified PI3Kα (p110α/p85α heterodimer) displays a Kₘ of approximately 1.8 μM for PI(4,5)P₂. In contrast, class III PI3K (Vps34) specifically phosphorylates phosphatidylinositol (PI) to produce PI(3)P, with a Kₘ of about 5 μM for PI, determined through similar radiolabeled lipid kinase assays on purified enzyme complexes. These kinetic parameters highlight the tuned substrate selectivity that enables precise spatiotemporal control of phosphoinositide signaling.[57][58] The catalytic activity of PI3Ks adheres to Michaelis-Menten kinetics. For the class IA isoform p110α, the maximum velocity (Vₘₐₓ) is approximately 0.5 nmol/min/mg under optimal conditions with PI(4,5)P₂ as substrate and excess ATP. This activity is potently inhibited by wortmannin, a fungal metabolite that covalently modifies a conserved lysine residue in the active site, yielding an IC₅₀ of 5 nM for class I PI3Ks. Such inhibition underscores the enzyme's reliance on precise active-site architecture for phosphate transfer.[57][58] The products of PI3K lipid kinase activity, particularly PI(3,4,5)P₃, mediate allosteric recruitment of downstream effectors bearing pleckstrin homology (PH) domains, such as protein kinase B (Akt/PKB), to cellular membranes. This localization facilitates Akt activation and propagation of survival signals. Additionally, negative feedback occurs through phosphatase and tensin homolog (PTEN), which dephosphorylates PI(3,4,5)P₃ at the D-3 position, reverting it to PI(4,5)P₂ and thereby attenuating signaling.[2]Protein Kinase Activity
Class I phosphoinositide 3-kinases (PI3Ks) exhibit protein kinase activity in addition to their well-characterized lipid kinase function, enabling direct phosphorylation of protein substrates on serine/threonine residues. This activity is mediated by the kinase domain of the p110 catalytic subunits, which utilizes ATP to transfer the γ-phosphate to target residues, distinct from the lipid-binding site involved in phosphoinositide phosphorylation. The protein kinase activity displays lower affinity for peptide substrates compared to lipids, with reported Km values in the range of tens to hundreds of micromolar, indicating a regulatory rather than primary catalytic role.[59][60] Evidence for this protein kinase function includes autophosphorylation of the p110 subunits themselves, primarily on C-terminal serine residues: Ser1070 in p110β, Ser1101 in p110γ, and Ser1039 in p110δ. Autophosphorylation of p110β and p110δ occurs in a Mn²⁺-dependent manner and down-regulates their lipid kinase activity, serving as a feedback mechanism, whereas p110γ autophosphorylation is Mg²⁺-dependent and enhanced by Gβγ subunits without inhibiting lipid catalysis. For p110α, autophosphorylation and phosphorylation of the associated p85 regulatory subunit on serine (e.g., Ser608) and tyrosine residues have been observed, with oncogenic mutants like H1047R exhibiting upregulated activity toward both p110 and p85α. These events are assayed in vitro using recombinant p110/p85 complexes incubated with [γ-³²P]ATP, followed by SDS-PAGE and autoradiography to detect phosphorylated bands, or by immunoblotting with phospho-specific antibodies.[60][14][59] Known protein substrates include the insulin receptor substrate-1 (IRS-1), where class I PI3K phosphorylates Ser/Thr residues, inhibiting IRS-1 tyrosine phosphorylation and disrupting insulin signaling as a negative feedback loop. This is evidenced by insulin-stimulated IRS-1 serine phosphorylation in cells, which is blocked by the PI3K inhibitor wortmannin, confirming the involvement of PI3K's protein kinase domain. Other substrates encompass the GM-CSF/IL-3 βc receptor (Ser585) and non-muscle tropomyosin 1α (Ser61), with p110α showing robust activity toward the βc fragment in kinase-dead mutant controls. For class IB p110γ, substrates include PKCζ, where phosphorylation activates downstream signaling in immune cells, though the precise sites remain under investigation. In vitro assays for these substrates often employ synthetic peptides or histone tails as proxies, revealing IC₅₀ values for peptide phosphorylation around 50 μM under physiological Mg²⁺ conditions.[61][59] Biologically, this protein kinase activity contributes to feedback regulation; for instance, IRS-1 phosphorylation by PI3K limits insulin pathway hyperactivity, while p110δ autophosphorylation in B cells modulates signaling thresholds for antigen receptor responses. Phosphorylation of p110δ enhances B-cell activation by fine-tuning PI3K output, as demonstrated in p110δ-deficient models showing impaired B-cell proliferation and survival upon BCR or CD40 stimulation. Overall, the protein kinase function complements lipid signaling by enabling direct modulation of key adaptors and receptors, with implications for metabolic and immune homeostasis.[61][14][62]Physiological Functions
Metabolic Regulation
Phosphoinositide 3-kinases (PI3Ks), particularly class IA isoforms, play a pivotal role in insulin signaling by integrating signals from the insulin receptor to regulate glucose homeostasis. Upon insulin binding to its receptor, the receptor tyrosine kinase phosphorylates insulin receptor substrates (IRS1 and IRS2), which recruit and activate class IA PI3K complexes composed of a p110 catalytic subunit and a p85 regulatory subunit. This activation leads to the phosphorylation of phosphatidylinositol-4,5-bisphosphate (PIP2) to produce phosphatidylinositol-3,4,5-trisphosphate (PIP3), a key second messenger that recruits phosphoinositide-dependent kinase 1 (PDK1) and protein kinase B (Akt) to the plasma membrane. Activated Akt then promotes glucose transporter 4 (GLUT4) translocation to the cell surface in muscle and adipose tissues, facilitating glucose uptake, while also inhibiting glycogen synthase kinase 3 (GSK3) to enhance glycogen synthesis in the liver.[63][64] Dysregulation of PI3K signaling contributes to metabolic disorders such as type 2 diabetes through impaired insulin sensitivity. Activating mutations in PIK3CA, encoding the p110α catalytic subunit, have been linked to insulin resistance by altering the balance of PI3K activity, leading to defective downstream signaling and reduced metabolic responses to insulin. Similarly, class III PI3K, represented by VPS34, is essential for autophagy-mediated nutrient sensing; VPS34 generates phosphatidylinositol-3-phosphate (PI3P) to initiate autophagosome formation, allowing cells to recycle nutrients during starvation and maintain energy balance. Disruptions in VPS34 activity impair this process, exacerbating metabolic stress in conditions like diabetes.[64][65] PI3K signaling intersects with energy balance pathways, including the mechanistic target of rapamycin complex 1 (mTORC1), which is indirectly activated via Akt to promote lipogenesis and anabolic processes in response to nutrient availability. Genetic studies in mice demonstrate this integration; for instance, tissue-specific knockouts of p110α (encoded by Pik3ca) in adipose tissue result in insulin resistance, lipodystrophy, and hyperglycemia due to impaired lipid storage and elevated circulating glucose levels. Elevated PIP3 levels have been correlated with features of metabolic syndrome, including obesity and dyslipidemia, reflecting hyperactive PI3K signaling that disrupts normal nutrient partitioning. Therapeutically, agents like metformin enhance PI3K-Akt signaling in hepatic tissues by upregulating IRS2 expression, thereby improving insulin sensitivity and glucose metabolism in insulin-resistant states.[64][66][67][68]Immune System Roles
Phosphoinositide 3-kinases (PI3Ks), particularly class I isoforms, play pivotal roles in both innate and adaptive immunity by generating phosphatidylinositol (3,4,5)-trisphosphate (PIP3), which serves as a second messenger to recruit and activate downstream effectors like Akt and PDK1 in immune cells.[69] Class IA PI3Ks, such as p110δ, are predominantly involved in antigen receptor signaling in lymphocytes, while class IB PI3K p110γ responds to G protein-coupled receptors (GPCRs) in myeloid cells and T cells, facilitating migration, activation, and effector functions.[70] These pathways ensure coordinated immune responses, from development and proliferation to pathogen clearance. In B-cell development and function, p110δ is essential, as it generates PIP3 upon B-cell receptor (BCR) engagement, leading to the recruitment of adaptor proteins like BLNK (B-cell linker) and subsequent activation of survival signals via Akt.[71] Deficiency in p110δ, as seen in knockout mice, results in reduced B-cell numbers, impaired BCR signaling, and defective antigen responses, culminating in immunodeficiency phenotypes such as decreased marginal zone and B1 B cells.[69] Gain-of-function mutations in PIK3CD (encoding p110δ) disrupt B-cell homeostasis, promoting excessive survival and contributing to immunodeficiencies like activated PI3Kδ syndrome (APDS).[72] For T-cell activation, p110γ mediates chemokine-induced responses through GPCRs, enabling T-cell migration and polarization toward inflammation sites by producing PIP3 that activates Rac and actin remodeling.[73] In parallel, class IA PI3Ks, including p110δ and p110α, support costimulatory signals from CD28 during T-cell receptor engagement, enhancing IL-2 production, proliferation, and differentiation into effector subsets like Th1 and Th17 cells.[74] This dual involvement ensures robust adaptive immunity, with p110γ knockout mice showing impaired T-cell chemotaxis and reduced immune responses to infections.[75] In innate immunity, p110γ is critical for neutrophil function, where it drives GPCR-dependent reactive oxygen species (ROS) production via NADPH oxidase assembly upon PIP3-mediated recruitment of p47phox and Rac.[76] Neutrophils from p110γ-deficient mice exhibit severely reduced ROS bursts in response to formyl peptides or complement, impairing bacterial killing and inflammation resolution.[77] Complementarily, class III PI3K (Vps34) regulates phagosome maturation by generating phosphatidylinositol 3-phosphate (PI3P), which recruits effectors like EEA1 for endosomal fusion and lysosomal delivery in macrophages and neutrophils.[78] Inhibition of Vps34 disrupts phagolysosome formation, highlighting its non-redundant role in innate pathogen clearance.[79] Links to autoimmunity underscore PI3K's regulatory balance; hyperactive p110δ signaling, as in APDS patients, drives lupus-like autoimmunity through excessive B- and T-cell activation, elevated autoantibodies, and lymphoproliferation mimicking systemic lupus erythematosus (SLE).[72] Conversely, Pik3cd knockout mice demonstrate reduced inflammation in autoimmune models, such as attenuated arthritis severity due to impaired T- and B-cell responses and lower cytokine production, indicating p110δ's promotion of pathogenic autoimmunity.[80] These findings position PI3K isoforms as key checkpoints in preventing immune dysregulation.[81]Cell Growth and Survival
Phosphoinositide 3-kinase (PI3K), particularly class I isoforms, plays a central role in regulating cell growth and survival in non-immune cells such as epithelial and fibroblastic lineages by transducing signals from receptor tyrosine kinases (RTKs). Upon activation by RTKs like the epidermal growth factor receptor (EGFR), class I PI3K generates phosphatidylinositol (3,4,5)-trisphosphate (PIP3) at the plasma membrane, which recruits and activates protein kinase B (Akt) through phosphoinositide-dependent kinase 1 (PDK1)-mediated phosphorylation.30865-6) Activated Akt then phosphorylates downstream targets, including the pro-apoptotic protein BAD at serine 136, sequestering it from Bcl-2 and preventing mitochondrial cytochrome c release to inhibit apoptosis.00248-4) Similarly, Akt phosphorylates forkhead box O (FOXO) transcription factors, excluding them from the nucleus and suppressing pro-apoptotic gene expression, thereby promoting cell survival and proliferation in response to growth factors.00248-4) In angiogenesis, class IA PI3K isoforms are essential for vascular endothelial growth factor (VEGF)-induced endothelial cell migration and vessel sprouting. VEGF binding to its receptor activates class IA PI3K, leading to PIP3 production that recruits Akt and Rac1 GTPase, facilitating actin cytoskeleton reorganization and directed migration of endothelial cells toward angiogenic cues.[82] This process is critical for new vessel formation in physiological contexts like tissue remodeling. Complementing this, class II PI3K isoforms, such as PI3K-C2α, support receptor recycling via clathrin-mediated endocytosis, ensuring sustained signaling from angiogenic receptors like VEGFR2. PI3K-C2α localizes to clathrin-coated pits, generating phosphatidylinositol 3-phosphate (PI3P) to promote endosomal maturation and receptor trafficking back to the plasma membrane, thereby amplifying migratory responses in endothelial cells.00191-5.pdf)[83] PI3K-C2α also contributes to wound healing by enhancing fibroblast motility and extracellular matrix remodeling. In fibroblasts, PI3K-C2α activation promotes lamellipodia formation and directed migration toward wound sites through localized PIP3 and PI3P production, facilitating re-epithelialization and tissue repair. Studies in PI3K-C2α knockout models reveal impaired fibroblast migration and delayed wound closure, with phenotypes showing reduced cell proliferation and adhesion at injury sites, underscoring its non-redundant role in cutaneous healing.[84][85] Negative feedback mechanisms within the PI3K pathway prevent excessive growth signaling and maintain cell cycle homeostasis. Ribosomal protein S6 kinase (S6K), downstream of mTORC1, phosphorylates insulin receptor substrate 1 (IRS1) at inhibitory sites like serine 302, reducing its association with PI3K and attenuating pathway activation to avoid overgrowth. This feedback loop is evident in quantitative models of PI3K flux during the cell cycle, where oscillatory PIP3 levels and S6K-mediated IRS1 inhibition synchronize growth signals with G1/S progression, ensuring balanced proliferation without unchecked expansion. Such dysregulation can parallel oncogenic contexts, but therapeutic targeting aims to restore this balance.[86][87][88]Neurological Functions
Phosphoinositide 3-kinase (PI3K) signaling plays a pivotal role in synaptic processes within the nervous system, particularly through specific isoforms that modulate long-term depression (LTD) and receptor dynamics. The class IB isoform p110γ, activated downstream of G protein-coupled receptors (GPCRs), is essential for GPCR-mediated LTD at synapses, facilitating homosynaptic NMDA receptor-dependent plasticity in hippocampal neurons. In parallel, class IA PI3K contributes to the trafficking of NMDA receptors to synaptic sites, enabling proper insertion and stabilization during synaptic remodeling in the hippocampus.00228-9) In the context of learning and memory, the PI3K-generated second messenger PIP3 activates the Akt pathway, which is critical for inducing long-term potentiation (LTP) in hippocampal CA1 synapses, a cellular correlate of spatial memory formation.[89] Conditional knockout of Pik3ca, encoding the p110α catalytic subunit of class IA PI3K, in astrocytes or neurons disrupts this pathway, leading to impairments in spatial memory tasks such as the Morris water maze. These findings underscore PI3K's isoform-specific contributions to cognitive functions, where disruptions alter synaptic strengthening without broadly affecting cell survival signals.[90] PI3K isoforms also support neuroprotection by regulating cellular maintenance and developmental processes. Class III PI3K, particularly Vps34, promotes autophagy in neurons, clearing protein aggregates and mitigating neurodegeneration in models of lysosomal storage disorders and aging-related decline.[91] Meanwhile, class II PI3K generates PI(3,4)P2 to facilitate axonal guidance during neuronal development, enabling growth cone navigation and proper circuit formation through microtubule stabilization.[92] Dysregulation of PI3K signaling previews neurological vulnerabilities, as hyperactivation—often via upstream mutations—promotes hyperexcitability and seizure susceptibility in epilepsy models by enhancing mTOR-dependent neuronal activity.[93] Similarly, PTEN loss, which removes negative regulation of the PI3K pathway, results in macrocephaly through excessive neuronal growth and dendritic hypertrophy in both human patients and mouse models.[94]Pathological Roles
Oncogenic Dysregulation in Cancer
Oncogenic dysregulation of phosphoinositide 3-kinase (PI3K) primarily involves genetic alterations that lead to hyperactivation of the pathway, promoting uncontrolled cell growth and survival in various malignancies. Hotspot mutations in the PIK3CA gene, encoding the p110α catalytic subunit of class I PI3K, are among the most common, with E542K and E545K in the helical domain and H1047R in the kinase domain occurring in approximately 30% of breast and colorectal cancers. These mutations enhance lipid kinase activity by 2- to 5-fold, resulting in elevated production of phosphatidylinositol (3,4,5)-trisphosphate (PIP3) and constitutive downstream signaling.[95][96][48] Amplification of the PIK3CB gene, encoding p110β, is frequently observed in prostate cancer, contributing to pathway activation independent of PIK3CA mutations. In glioblastoma, loss of PTEN, a key negative regulator that dephosphorylates PIP3, leads to unchecked PI3K signaling and is present in up to 40% of cases. These alterations sustain PIP3-mediated activation of Akt and mTOR, driving tumor cell proliferation, survival, and invasion; while class I PI3Ks dominate in primary tumor initiation, class II and III isoforms have emerging roles in facilitating metastasis through regulation of vesicular trafficking and cytoskeletal dynamics.[97][98][99] Epidemiologically, alterations in the PI3K pathway, including mutations, amplifications, and upstream/downstream changes, are present in approximately 44% of solid tumors, as evidenced by comprehensive analyses of over 60,000 samples.[100] The Cancer Genome Atlas (TCGA) data reveal frequent co-occurrence of PI3K pathway mutations with KRAS alterations in cancers such as colorectal and pancreatic, potentially amplifying oncogenic signaling through parallel MAPK pathway activation.[101]Involvement in Inflammation and Autoimmunity
Phosphoinositide 3-kinase (PI3K) isoforms, particularly p110δ and p110γ, play critical pro-inflammatory roles in rheumatoid arthritis (RA) through hyperactivation that drives immune cell dysfunction and joint pathology. In RA, elevated expression of p110δ in synovial fibroblasts and T cells promotes aberrant cell survival, migration, and cytokine secretion, exacerbating synovitis and cartilage erosion.[102] Similarly, p110γ hyperactivation in neutrophils and macrophages facilitates their recruitment to inflamed joints, amplifying tissue damage via reactive oxygen species and protease release.[103] Inhibition of these isoforms in murine models, such as collagen-induced arthritis, significantly reduces paw swelling, neutrophil infiltration, and bone erosion, underscoring their pathogenic contributions.[104] PI3K signaling via p110δ is essential for Th17 cell differentiation and effector function in RA, where it enhances IL-6- and TGF-β-mediated STAT3 activation, leading to increased IL-17 production that perpetuates chronic inflammation.[70] Hyperactive p110δ also sustains TNFα secretion from synovial T cells and macrophages, forming a feed-forward loop that recruits additional inflammatory cells and promotes osteoclast activation.[105] Dual inhibition of p110δ and p110γ in RA models suppresses Th17 responses and cytokine storms more effectively than single-isoform blockade, highlighting their synergistic roles in driving pro-inflammatory T cell polarization.[104] In autoimmunity, gain-of-function mutations in PIK3CD, encoding p110δ, cause activated PI3K delta syndrome (APDS), a primary immunodeficiency characterized by dysregulated immune activation. These mutations lead to constitutive PI3Kδ hyperactivity, resulting in excessive B and T cell proliferation, lymphadenopathy, and splenomegaly due to impaired apoptosis and enhanced survival signaling via PIP3/Akt.[106] APDS patients exhibit lymphoproliferation alongside autoimmune cytopenias and enteropathy, stemming from failed immune tolerance and autoreactive lymphocyte expansion.[106] The syndrome also predisposes to recurrent sinopulmonary infections from defective humoral immunity and impaired neutrophil function, illustrating how PI3Kδ gain-of-function disrupts immune homeostasis.[106] PI3K-mediated feedback loops amplify inflammation in macrophages, where PIP3 accumulation sustains NF-κB activation to prolong pro-inflammatory gene expression. In response to TLR ligands, class I PI3K generates PIP3, which recruits Akt to phosphorylate and inhibit GSK3, thereby stabilizing IκB kinase and enabling sustained NF-κB nuclear translocation for cytokine transcription.[107] This positive feedback exacerbates chronic inflammation in autoimmune settings by maintaining macrophage polarization toward an M1 phenotype. Conversely, class III PI3K (Vps34) deficiency disrupts autophagy and endosomal trafficking, worsening colitis in experimental models through unchecked bacterial invasion and epithelial barrier breakdown.[108] In Vps34-deficient mice and zebrafish, intestinal inflammation intensifies with increased cytokine release and immune cell infiltration, linking PI3K class III to mucosal homeostasis. Clinical correlations in systemic lupus erythematosus (SLE) reveal elevated PI3K activity as a hallmark of disease progression, particularly in renal and lymphoid tissues. In SLE patients and murine models, hyperactive PI3K/Akt/mTOR signaling in B cells and T cells drives autoantibody production and immune complex deposition, correlating with lupus nephritis severity.[109] Increased p110δ expression and downstream phosphorylation (e.g., of Akt at Ser473) serve as potential biomarkers, with higher levels in peripheral blood mononuclear cells associating with disease flares and reduced response to standard therapies.[110] Targeting this pathway in SLE models ameliorates glomerulonephritis and autoimmunity, supporting PI3Kδ as a therapeutic node.[110]Effects on Neurological and Cardiovascular Disorders
Dysregulation of phosphoinositide 3-kinase (PI3K) signaling plays a significant role in neurological disorders, particularly through somatic mutations in the PIK3CA gene, which encodes the p110α catalytic subunit of class IA PI3K. These mutations lead to hyperactivation of the PI3K/AKT/mTOR pathway, causing brain overgrowth malformations such as hemimegalencephaly, a condition characterized by enlargement of one cerebral hemisphere often accompanied by severe, drug-resistant epilepsy. In affected individuals, mosaic PIK3CA mutations disrupt normal neuronal proliferation and migration, resulting in cortical dysplasia and epileptogenic foci that manifest as early-onset seizures. Studies in mouse models harboring these mutations recapitulate the phenotype, showing enlarged brain regions, fluid accumulation, and spontaneous epileptic activity, underscoring the pathway's causal role in these epileptogenic malformations.[111][112] In Alzheimer's disease, class I PI3K signaling is implicated in tau pathology via the downstream AKT/GSK3β axis, where impaired pathway activity contributes to tau hyperphosphorylation. Normally, PI3K generates PIP3 to activate AKT, which inhibits GSK3β and prevents excessive phosphorylation of tau at sites like Ser396 and Thr231; however, in Alzheimer's, amyloid-β accumulation and presenilin-1 mutations disrupt this activation, leading to GSK3β hyperactivity and tau aggregation into neurofibrillary tangles. This dysregulation exacerbates neuronal loss and cognitive decline, as evidenced in APP/PS1 mouse models where restoring PI3K/AKT activity reduces tau hyperphosphorylation and improves synaptic function. Additionally, reduced PIP3-AKT signaling promotes amyloid-β toxicity by failing to suppress pro-apoptotic pathways, allowing Aβ to induce synaptic dysfunction and oxidative stress in neurons.[113][114][115] PI3K involvement extends to stroke recovery, where class I isoforms support neuroprotection and plasticity in the post-ischemic brain. Recent studies from the 2020s demonstrate that PI3K/AKT activation enhances axonal outgrowth, reduces inflammation, and promotes functional recovery in rodent models of ischemic stroke, such as through upregulation of miR-29a targeting PTEN to boost pathway activity. In global cerebral ischemia-reperfusion models, pharmacological PI3K agonists like alisol A 24-acetate mitigate neuronal apoptosis and improve behavioral outcomes by modulating AKT-dependent anti-inflammatory effects. These findings highlight PI3K as a therapeutic target for enhancing rehabilitation after stroke, contrasting its normal role in neuronal survival and briefly linking to baseline neurological functions like synaptic maintenance.[116][117][118] In cardiovascular disorders, class IA PI3K isoforms, particularly p110α, contribute to pathological cardiac hypertrophy triggered by pressure overload, such as in aortic constriction models. Activation of this pathway initially supports adaptive remodeling but, when sustained, drives maladaptive hypertrophy, interstitial fibrosis, and contractile dysfunction through excessive AKT/mTOR signaling that promotes cardiomyocyte enlargement without preserving function. Genetic studies show that cardiac-specific p110α overexpression exacerbates fibrosis and heart failure progression under pressure overload, while balanced activation via exercise preconditioning protects against these outcomes by limiting pathological extracellular matrix deposition.[119][120][121] Class III PI3K, known as Vps34, regulates autophagy in cardiomyocytes, and its dysregulation contributes to heart failure by impairing autophagic flux. Prolonged activation of Vps34 generates PI3P to initiate excessive autophagy, which transitions compensatory hypertrophy to decompensated failure in pressure-overloaded hearts, leading to protein degradation and energy deficits. In mouse models of transverse aortic constriction, Vps34 inhibition restores autophagic balance, reduces infarct size, and preserves ejection fraction, demonstrating its essential yet detrimental role when overactive in failing hearts.[122][123] Regarding arrhythmias, inhibition of the class IB isoform p110γ has shown protective effects in mouse models of catecholamine-induced ventricular tachyarrhythmias. p110γ-null mice exhibit altered cAMP regulation and repolarization, but selective pharmacological inhibition with compounds like AS605240 reduces susceptibility to β-adrenergic-triggered arrhythmias by dampening inflammatory signaling and calcium overload in stressed cardiomyocytes. This suggests targeted p110γ blockade as a strategy to mitigate arrhythmia risk in hypertensive or ischemic hearts, building on its role in modulating cardiac excitability.[124][125]Therapeutic Inhibition
Inhibitor Classes and Mechanisms
Phosphoinositide 3-kinase (PI3K) inhibitors are classified primarily based on their selectivity for specific isoforms or classes, as well as their binding modes, which influence potency, specificity, and potential off-target effects. Isoform-specific inhibitors target individual catalytic subunits of class I PI3Ks, such as p110δ, to minimize toxicity associated with pan-inhibition, while pan-PI3K inhibitors affect multiple class I isoforms (p110α, β, δ, γ). For example, idelalisib is a selective p110δ inhibitor that acts as a covalent ATP-competitive agent, binding irreversibly to a cysteine residue in the ATP-binding pocket, with an IC50 of 2.5 nM for p110δ and over 40-fold selectivity against other class I isoforms.[126] In contrast, copanlisib is a reversible pan-class I PI3K inhibitor that binds competitively to the ATP site across all four isoforms, exhibiting subnanomolar potency against p110α and δ while maintaining micromolar activity against p110β and γ.[127] Inhibitors can also be distinguished by orthosteric (ATP-site) versus allosteric binding mechanisms, with the latter targeting sites outside the conserved ATP pocket, such as the helical domain, to achieve greater isoform selectivity. Duvelisib exemplifies orthosteric inhibition as a dual p110δ/γ inhibitor that reversibly binds the ATP site, showing IC50 values of 2.4 nM and 0.1 nM for these isoforms, respectively, and sparing p110α and β.[128] Allosteric inhibitors, such as those targeting the helical domain of p110β (e.g., SAR260301), bind to a non-ATP site to selectively modulate isoform activity; SAR260301 demonstrates an IC50 of 23 nM for p110β with over 60-fold selectivity against other class I isoforms, though its development was limited by rapid clearance.[129] Dual inhibitors target PI3K alongside downstream or related kinases like mTOR to block compensatory pathway activation. Dactolisib (NVP-BEZ235) is an ATP-competitive dual PI3K/mTOR inhibitor that potently suppresses class I PI3K isoforms (IC50 ~5-20 nM) and mTORC1/2 (IC50 ~100 nM), preventing feedback reactivation of Akt.[130] For class III PI3K (Vps34), inhibitors like 3-methyladenine (3-MA) provide non-specific blockade, acting as a weak, reversible inhibitor with an IC50 of approximately 25 μM for Vps34 but also affecting class I PI3Ks at higher concentrations, limiting its selectivity.[131] Resistance to PI3K inhibitors often arises from mutations that alter binding affinity or pathway reactivation. The E545K mutation in the helical domain of p110α reduces inhibitor binding to the ATP site, thereby decreasing sensitivity to isoform-specific agents like idelalisib in cells reliant on p110α activity, with up to 10-fold shifts in IC50 observed in mutant-expressing models.[132] Pharmacokinetic properties, such as oral bioavailability, further impact clinical utility; idelalisib exhibits high oral bioavailability (>70% in preclinical models), enabling once-daily dosing, whereas copanlisib's lower oral absorption (~30%) favors intravenous administration to achieve therapeutic exposure.[126] Duvelisib also demonstrates favorable oral bioavailability (~50-60%), supporting its systemic inhibition profile.[128]| Inhibitor | Type | Binding Mechanism | Key Selectivity (IC50) | Oral Bioavailability |
|---|---|---|---|---|
| Idelalisib | Isoform-specific (p110δ) | Covalent ATP-competitive | 2.5 nM (p110δ) | High (>70%)[126] |
| Copanlisib | Pan-class I | Reversible ATP-competitive | <1 nM (p110α/δ) | Moderate (~30%)[127] |
| Duvelisib | Dual (p110δ/γ) | Reversible ATP-competitive | 2.4 nM (δ), 0.1 nM (γ) | Favorable (~50-60%)[128] |
| SAR260301 | Isoform-specific (p110β) | Allosteric (helical domain) | 23 nM (p110β) | Limited (rapid clearance)[129] |
| Dactolisib | Dual PI3K/mTOR | ATP-competitive | ~5-20 nM (class I PI3K) | High[130] |
| 3-MA | Class III (Vps34) | Reversible, non-specific | ~25 μM (Vps34) | N/A (research tool)[131] |