Community hub
Recent from talks
Knowledge base stats:
Talk channels stats:
Members stats:
Sanger sequencing
Sanger sequencing is a method of DNA sequencing that involves electrophoresis and is based on the random incorporation of chain-terminating dideoxynucleotides by DNA polymerase during in vitro DNA replication. After first being developed by Frederick Sanger and colleagues in 1977, it became the most widely used sequencing method for approximately 40 years. An automated instrument using slab gel electrophoresis and fluorescent labels was first commercialized by Applied Biosystems in March 1987. Later, automated slab gels were replaced with automated capillary array electrophoresis.
Recently, higher volume Sanger sequencing has been replaced by next generation sequencing methods, especially for large-scale, automated genome analyses. However, the Sanger method remains in wide use for smaller-scale projects and for validation of deep sequencing results. It still has the advantage over short-read sequencing technologies (like Illumina) in that it can produce DNA sequence reads of > 500 nucleotides and maintains a very low error rate with accuracies around 99.99%. Sanger sequencing is still actively being used in efforts for public health initiatives such as sequencing the spike protein from SARS-CoV-2 as well as for the surveillance of norovirus outbreaks through the United States Center for Disease Control and Prevention (CDC)'s CaliciNet surveillance network.
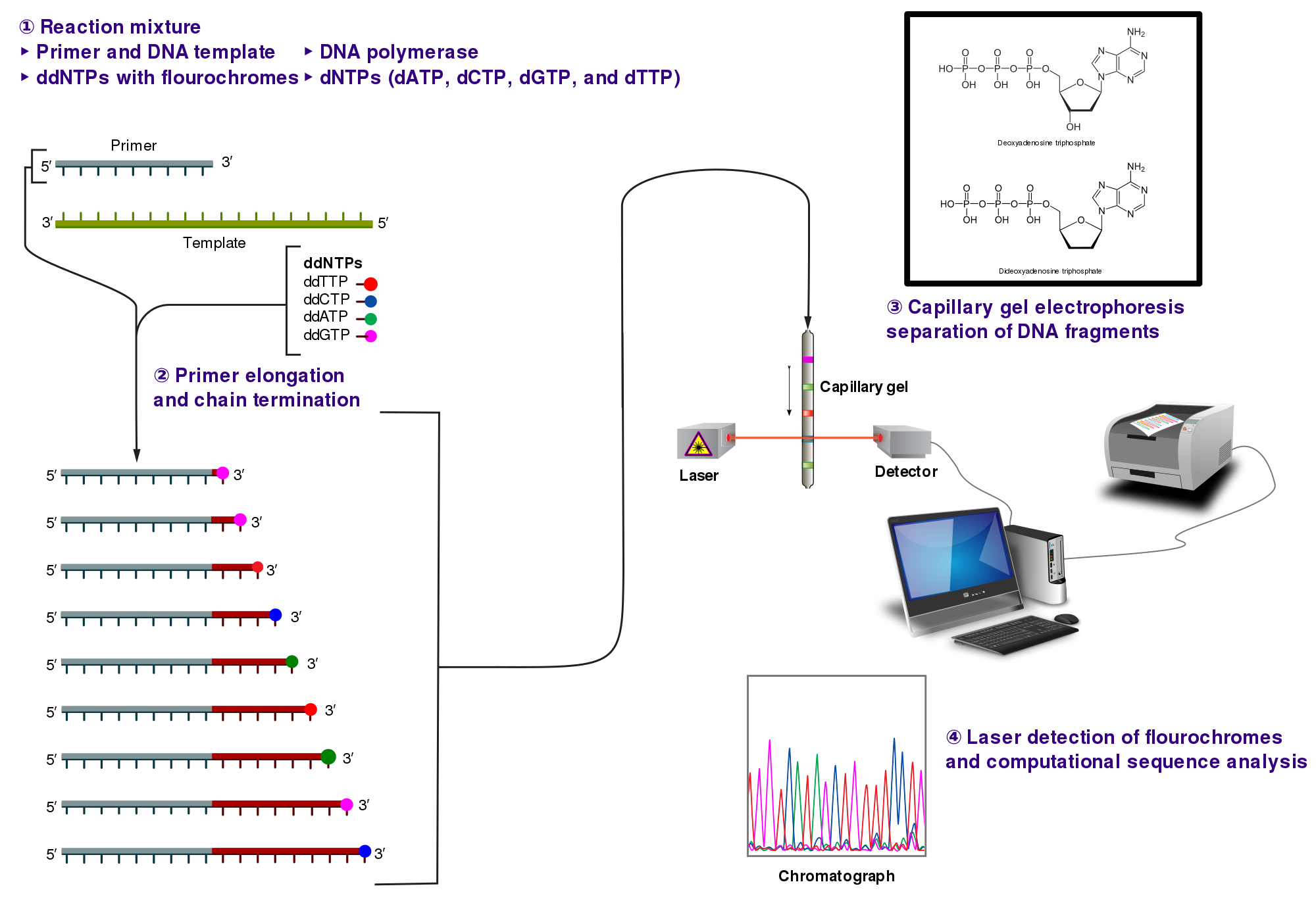
The classical chain-termination method requires a single-stranded DNA template, a DNA primer, a DNA polymerase, normal deoxynucleotide triphosphates (dNTPs), and modified di-deoxynucleotide triphosphates (ddNTPs), the latter of which terminate DNA strand elongation. These chain-terminating nucleotides lack a 3'-OH group required for the formation of a phosphodiester bond between two nucleotides, causing DNA polymerase to cease extension of DNA when a modified ddNTP is incorporated. The ddNTPs may be radioactively or fluorescently labelled for detection in automated sequencing machines.
The DNA sample is divided into four separate sequencing reactions, containing all four of the standard deoxynucleotides (dATP, dGTP, dCTP and dTTP) and the DNA polymerase. To each reaction is added only one of the four dideoxynucleotides (ddATP, ddGTP, ddCTP, or ddTTP), while the other added nucleotides are ordinary ones. The deoxynucleotide concentration should be approximately 100-fold higher than that of the corresponding dideoxynucleotide (e.g. 0.5mM dTTP : 0.005mM ddTTP) to allow enough fragments to be produced while still transcribing the complete sequence (but the concentration of ddNTP also depends on the desired length of sequence). Putting it in a more sensible order, four separate reactions are needed in this process to test all four ddNTPs. Following rounds of template DNA extension from the bound primer, the resulting DNA fragments are heat denatured and separated by size using gel electrophoresis. In the original publication of 1977, the formation of base-paired loops of ssDNA was a cause of serious difficulty in resolving bands at some locations. This is frequently performed using a denaturing polyacrylamide-urea gel with each of the four reactions run in one of four individual lanes (lanes A, T, G, C). The DNA bands may then be visualized by autoradiography or UV light, and the DNA sequence can be directly read off the X-ray film or gel image.
In the image on the right, X-ray film was exposed to the gel, and the dark bands correspond to DNA fragments of different lengths. A dark band in a lane indicates a DNA fragment that is the result of chain termination after incorporation of a dideoxynucleotide (ddATP, ddGTP, ddCTP, or ddTTP). The relative positions of the different bands among the four lanes, from bottom to top, are then used to read the DNA sequence.
Technical variations of chain-termination sequencing include tagging with nucleotides containing radioactive phosphorus for radiolabelling, or using a primer labeled at the 5' end with a fluorescent dye. Dye-primer sequencing facilitates reading in an optical system for faster and more economical analysis and automation. The later development by Leroy Hood and coworkers of fluorescently labeled ddNTPs and primers set the stage for automated, high-throughput DNA sequencing.
Chain-termination methods have greatly simplified DNA sequencing. For example, chain-termination-based kits are commercially available that contain the reagents needed for sequencing, pre-aliquoted and ready to use. Limitations include non-specific binding of the primer to the DNA, affecting accurate read-out of the DNA sequence, and DNA secondary structures affecting the fidelity of the sequence.
Dye-terminator sequencing utilizes labelling of the chain terminator ddNTPs, which permits sequencing in a single reaction rather than four reactions as in the labelled-primer method. In dye-terminator sequencing, each of the four dideoxynucleotide chain terminators is labelled with fluorescent dyes, each of which emits light at different wavelengths.
Hub AI
Sanger sequencing AI simulator
(@Sanger sequencing_simulator)
Sanger sequencing
Sanger sequencing is a method of DNA sequencing that involves electrophoresis and is based on the random incorporation of chain-terminating dideoxynucleotides by DNA polymerase during in vitro DNA replication. After first being developed by Frederick Sanger and colleagues in 1977, it became the most widely used sequencing method for approximately 40 years. An automated instrument using slab gel electrophoresis and fluorescent labels was first commercialized by Applied Biosystems in March 1987. Later, automated slab gels were replaced with automated capillary array electrophoresis.
Recently, higher volume Sanger sequencing has been replaced by next generation sequencing methods, especially for large-scale, automated genome analyses. However, the Sanger method remains in wide use for smaller-scale projects and for validation of deep sequencing results. It still has the advantage over short-read sequencing technologies (like Illumina) in that it can produce DNA sequence reads of > 500 nucleotides and maintains a very low error rate with accuracies around 99.99%. Sanger sequencing is still actively being used in efforts for public health initiatives such as sequencing the spike protein from SARS-CoV-2 as well as for the surveillance of norovirus outbreaks through the United States Center for Disease Control and Prevention (CDC)'s CaliciNet surveillance network.
The classical chain-termination method requires a single-stranded DNA template, a DNA primer, a DNA polymerase, normal deoxynucleotide triphosphates (dNTPs), and modified di-deoxynucleotide triphosphates (ddNTPs), the latter of which terminate DNA strand elongation. These chain-terminating nucleotides lack a 3'-OH group required for the formation of a phosphodiester bond between two nucleotides, causing DNA polymerase to cease extension of DNA when a modified ddNTP is incorporated. The ddNTPs may be radioactively or fluorescently labelled for detection in automated sequencing machines.
The DNA sample is divided into four separate sequencing reactions, containing all four of the standard deoxynucleotides (dATP, dGTP, dCTP and dTTP) and the DNA polymerase. To each reaction is added only one of the four dideoxynucleotides (ddATP, ddGTP, ddCTP, or ddTTP), while the other added nucleotides are ordinary ones. The deoxynucleotide concentration should be approximately 100-fold higher than that of the corresponding dideoxynucleotide (e.g. 0.5mM dTTP : 0.005mM ddTTP) to allow enough fragments to be produced while still transcribing the complete sequence (but the concentration of ddNTP also depends on the desired length of sequence). Putting it in a more sensible order, four separate reactions are needed in this process to test all four ddNTPs. Following rounds of template DNA extension from the bound primer, the resulting DNA fragments are heat denatured and separated by size using gel electrophoresis. In the original publication of 1977, the formation of base-paired loops of ssDNA was a cause of serious difficulty in resolving bands at some locations. This is frequently performed using a denaturing polyacrylamide-urea gel with each of the four reactions run in one of four individual lanes (lanes A, T, G, C). The DNA bands may then be visualized by autoradiography or UV light, and the DNA sequence can be directly read off the X-ray film or gel image.
In the image on the right, X-ray film was exposed to the gel, and the dark bands correspond to DNA fragments of different lengths. A dark band in a lane indicates a DNA fragment that is the result of chain termination after incorporation of a dideoxynucleotide (ddATP, ddGTP, ddCTP, or ddTTP). The relative positions of the different bands among the four lanes, from bottom to top, are then used to read the DNA sequence.
Technical variations of chain-termination sequencing include tagging with nucleotides containing radioactive phosphorus for radiolabelling, or using a primer labeled at the 5' end with a fluorescent dye. Dye-primer sequencing facilitates reading in an optical system for faster and more economical analysis and automation. The later development by Leroy Hood and coworkers of fluorescently labeled ddNTPs and primers set the stage for automated, high-throughput DNA sequencing.
Chain-termination methods have greatly simplified DNA sequencing. For example, chain-termination-based kits are commercially available that contain the reagents needed for sequencing, pre-aliquoted and ready to use. Limitations include non-specific binding of the primer to the DNA, affecting accurate read-out of the DNA sequence, and DNA secondary structures affecting the fidelity of the sequence.
Dye-terminator sequencing utilizes labelling of the chain terminator ddNTPs, which permits sequencing in a single reaction rather than four reactions as in the labelled-primer method. In dye-terminator sequencing, each of the four dideoxynucleotide chain terminators is labelled with fluorescent dyes, each of which emits light at different wavelengths.