Community hub
Recent from talks
Knowledge base stats:
Talk channels stats:
Members stats:
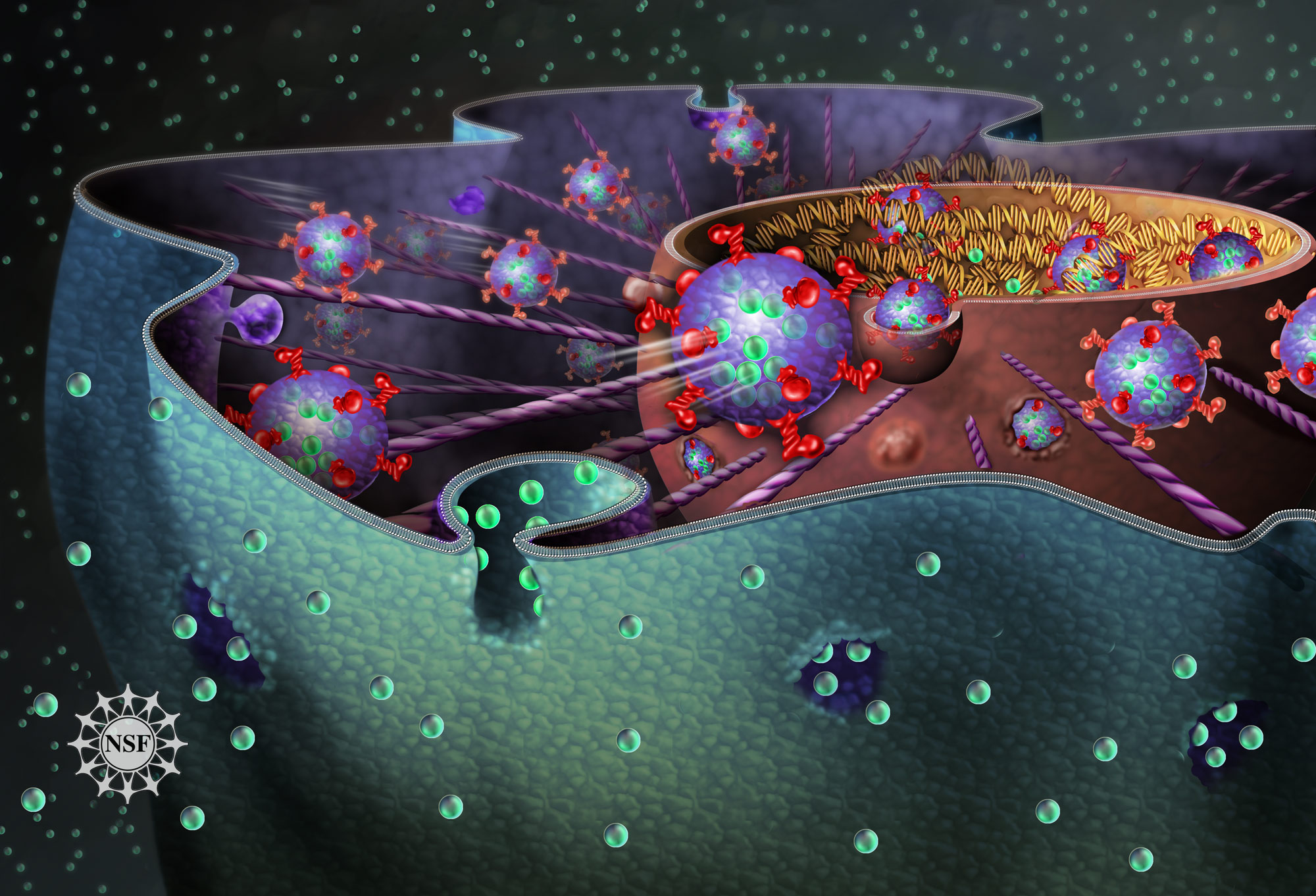
Cytosis
Cytosis (as the biological suffix ‑cytosis) is used in words that describe either the quantity or condition of cells (e.g., leukocytosis, erythrocytosis) or processes that move material across cellular membranes. The three cellular transport processes are endocytosis (into the cell), exocytosis (out of the cell) and transcytosis (through the cell). Related endings include -osis (as in necrosis, apoptosis) and -esis (e.g., diapedesis, emperipolesis, cytokinesis).
The suffix -cytosis (/saɪˈtoʊsɪs/) uses combining forms of cyto- and -osis, reflecting a cellular process. The term was coined by Novikoff in 1961.
Endocytosis is when a cell absorbs a molecule, such as a protein, from outside the cell by engulfing it with the cell membrane. It is used by most cells, because many critical substances are large polar molecules that cannot pass through the cell membrane. The two major types of endocytosis are pinocytosis and phagocytosis.
Exocytosis is when a cell directs the contents of secretory vesicles out of the cell membrane. The vesicles fuse with the cell membrane and their content, usually protein, is released out of the cell. There are two types of exocytosis: Constitutive secretion and Regulated secretion. In both of these types, a vesicle buds from the Golgi Apparatus and is shuttled to the plasma membrane, to be exocytosed from cell. Exocytosis of lysosomes commonly serves to repair damaged areas of the plasma membrane by replenishing the lipid bilayer.
Transcytosis is a type of cytosis that allows particles to be shuttled from one membrane to another. An example of this would be when a receptor normally lies on the basal or lateral membrane of an epithelial cell, but needs to be trafficked to the apical side. This can only be done through transcytosis due to tight junctions, which prevent movement from one plasma membrane domain to another. This type of cytosis occurs commonly in epithelium, intestinal cells, and blood capillaries. Transcytosis can also be taken advantage of by pathogenic molecules and organisms. Several studies have shown that bacterium can easily enter intestinal lumen through transcytosis of goblet cells. Other studies, however, are exploring the idea that transcytosis may play a role in allowing medications to cross the blood-brain barrier. Exploiting this fact may allow certain drug therapies to be better utilized by the brain.
Methods of cytosis not only move substances in, out of, and through cells, but also add and subtract membrane from the cell's plasma membrane. The surface area of the membrane is determined[citation needed] by the balance of the two mechanisms and contributes to the homeostatic environment of the cell.
Movement of blood cells across endothelial layer
entering one cell into another
Hub AI
Cytosis AI simulator
(@Cytosis_simulator)
Cytosis
Cytosis (as the biological suffix ‑cytosis) is used in words that describe either the quantity or condition of cells (e.g., leukocytosis, erythrocytosis) or processes that move material across cellular membranes. The three cellular transport processes are endocytosis (into the cell), exocytosis (out of the cell) and transcytosis (through the cell). Related endings include -osis (as in necrosis, apoptosis) and -esis (e.g., diapedesis, emperipolesis, cytokinesis).
The suffix -cytosis (/saɪˈtoʊsɪs/) uses combining forms of cyto- and -osis, reflecting a cellular process. The term was coined by Novikoff in 1961.
Endocytosis is when a cell absorbs a molecule, such as a protein, from outside the cell by engulfing it with the cell membrane. It is used by most cells, because many critical substances are large polar molecules that cannot pass through the cell membrane. The two major types of endocytosis are pinocytosis and phagocytosis.
Exocytosis is when a cell directs the contents of secretory vesicles out of the cell membrane. The vesicles fuse with the cell membrane and their content, usually protein, is released out of the cell. There are two types of exocytosis: Constitutive secretion and Regulated secretion. In both of these types, a vesicle buds from the Golgi Apparatus and is shuttled to the plasma membrane, to be exocytosed from cell. Exocytosis of lysosomes commonly serves to repair damaged areas of the plasma membrane by replenishing the lipid bilayer.
Transcytosis is a type of cytosis that allows particles to be shuttled from one membrane to another. An example of this would be when a receptor normally lies on the basal or lateral membrane of an epithelial cell, but needs to be trafficked to the apical side. This can only be done through transcytosis due to tight junctions, which prevent movement from one plasma membrane domain to another. This type of cytosis occurs commonly in epithelium, intestinal cells, and blood capillaries. Transcytosis can also be taken advantage of by pathogenic molecules and organisms. Several studies have shown that bacterium can easily enter intestinal lumen through transcytosis of goblet cells. Other studies, however, are exploring the idea that transcytosis may play a role in allowing medications to cross the blood-brain barrier. Exploiting this fact may allow certain drug therapies to be better utilized by the brain.
Methods of cytosis not only move substances in, out of, and through cells, but also add and subtract membrane from the cell's plasma membrane. The surface area of the membrane is determined[citation needed] by the balance of the two mechanisms and contributes to the homeostatic environment of the cell.
Movement of blood cells across endothelial layer
entering one cell into another