
Community hub
Recent from talks
Contribute something to knowledge base
Content stats: 0 posts, 0 articles, 1 media, 0 notes
Members stats: 0 subscribers, 0 contributors, 0 moderators, 0 supporters
Subscribers
Supporters
Contributors
Moderators
Hub AI
Daratumumab AI simulator
(@Daratumumab_simulator)
Hub AI
Daratumumab AI simulator
(@Daratumumab_simulator)
Daratumumab
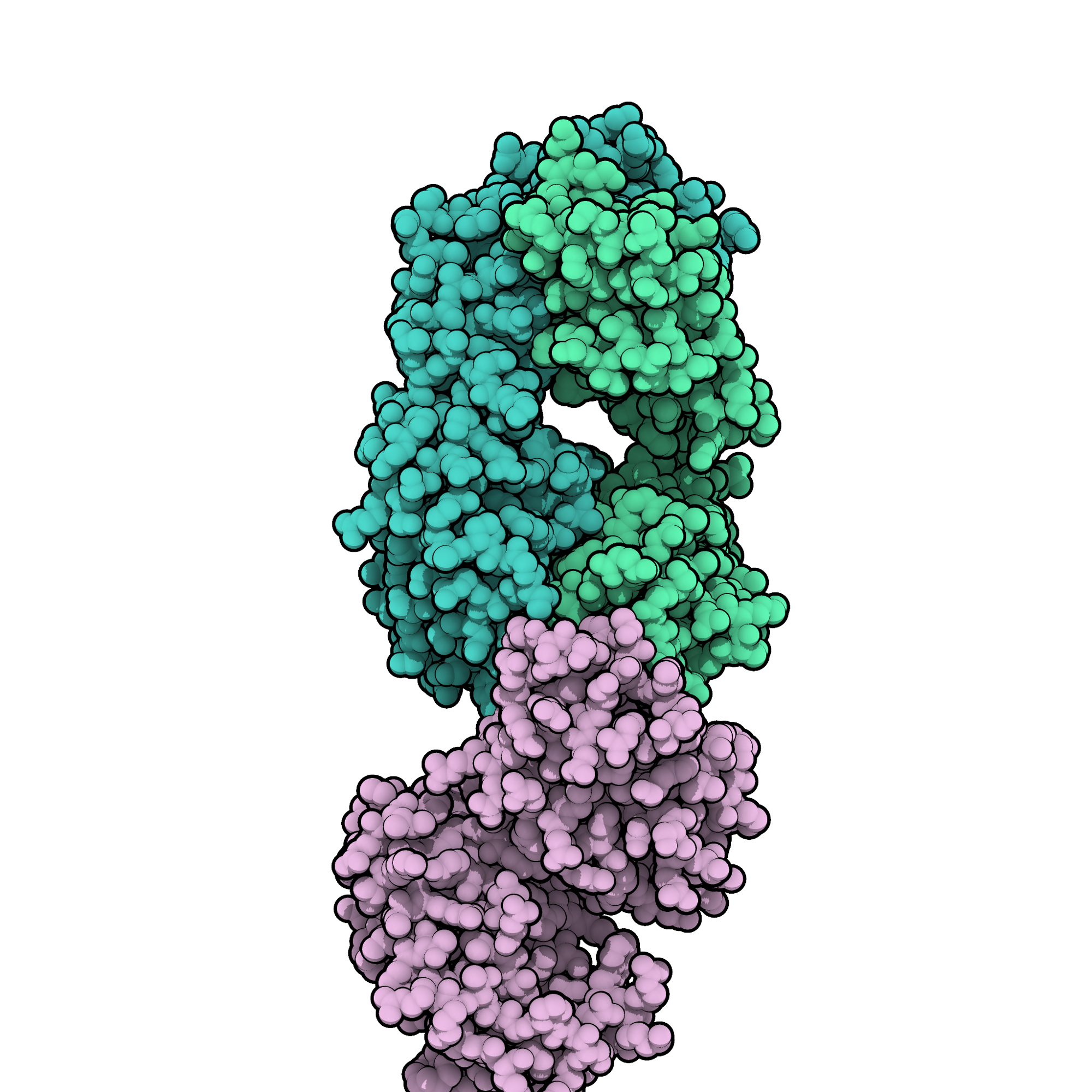
Daratumumab, sold under the brand name Darzalex among others, is an anti-cancer monoclonal antibody medication. It binds to CD38, which is overexpressed in multiple myeloma cells. Daratumumab was originally developed by Genmab, but it is now being jointly developed by Genmab along with the Johnson & Johnson subsidiary Janssen Biotech, which acquired worldwide commercialization rights to the drug from Genmab.
Daratumumab was granted breakthrough therapy drug status in 2013, for multiple myeloma. It was granted orphan drug status for multiple myeloma, diffuse large B cell lymphoma, follicular lymphoma, and mantle cell lymphoma.
It is available in combination with hyaluronidase as daratumumab/hyaluronidase (brand name Darzalex Faspro).
In May 2018, the US Food and Drug Administration (FDA) approved daratumumab for use in combination with bortezomib, melphalan and prednisone to include the treatment of people with newly diagnosed multiple myeloma who are ineligible for autologous stem cell transplant.
In the European Union it is indicated as monotherapy for the treatment of adults with relapsed and refractory multiple myeloma, whose prior therapy included a proteasome inhibitor and an immunomodulatory agent and who have demonstrated disease progression on the last therapy.
Treatment of multiple myeloma with daratumumab potentially increases the patient's susceptibility to bacterial and viral infections, due to the killing of natural killer cells (which are the main innate immune system defense against virus). Daratumumab frequently causes human cytomegalovirus (CMV) reactivation by an unknown mechanism. Injection related reactions (inflammation-like) are also common.
Daratumumab can also bind to CD38 present on red blood cells and interfere with routine testing for clinically significant antibodies. People will show a panel-reactive antibody response, including a positive auto-control, which tends to mask the presence of any clinically significant antibodies. Treatment of the antibody panel cells with dithiothreitol (DTT) and repeating testing will effectively negate the binding of daratumumab to CD38 on the red blood cell surface; however, DTT also inactivates/destroys many antigens on the red blood cell surface by disrupting disulfide bonds. The only antigen system affected that is associated with common, clinically significant antibodies is Kell, making crossmatch testing with K-negative RBCs a reasonable alternative when urgent transfusion is indicated. It is therefore advisable to do a baseline antibody screen and Rh & Kell phenotyping (type and screen) before starting the therapy. If antibody screen is negative, proceed with phenotype matched transfusions during therapy. If antibody screen is positive, give specific antigen negative blood. The incompatibility may persist for up to 6 months after stopping the medicine. Furthermore, blood transfusion centers should be routinely notified when sending such a sample.
Daratumumab can also interfere with flow cytometric evaluation of multiple myeloma, causing an apparent lack of plasma cells.
Daratumumab
Daratumumab, sold under the brand name Darzalex among others, is an anti-cancer monoclonal antibody medication. It binds to CD38, which is overexpressed in multiple myeloma cells. Daratumumab was originally developed by Genmab, but it is now being jointly developed by Genmab along with the Johnson & Johnson subsidiary Janssen Biotech, which acquired worldwide commercialization rights to the drug from Genmab.
Daratumumab was granted breakthrough therapy drug status in 2013, for multiple myeloma. It was granted orphan drug status for multiple myeloma, diffuse large B cell lymphoma, follicular lymphoma, and mantle cell lymphoma.
It is available in combination with hyaluronidase as daratumumab/hyaluronidase (brand name Darzalex Faspro).
In May 2018, the US Food and Drug Administration (FDA) approved daratumumab for use in combination with bortezomib, melphalan and prednisone to include the treatment of people with newly diagnosed multiple myeloma who are ineligible for autologous stem cell transplant.
In the European Union it is indicated as monotherapy for the treatment of adults with relapsed and refractory multiple myeloma, whose prior therapy included a proteasome inhibitor and an immunomodulatory agent and who have demonstrated disease progression on the last therapy.
Treatment of multiple myeloma with daratumumab potentially increases the patient's susceptibility to bacterial and viral infections, due to the killing of natural killer cells (which are the main innate immune system defense against virus). Daratumumab frequently causes human cytomegalovirus (CMV) reactivation by an unknown mechanism. Injection related reactions (inflammation-like) are also common.
Daratumumab can also bind to CD38 present on red blood cells and interfere with routine testing for clinically significant antibodies. People will show a panel-reactive antibody response, including a positive auto-control, which tends to mask the presence of any clinically significant antibodies. Treatment of the antibody panel cells with dithiothreitol (DTT) and repeating testing will effectively negate the binding of daratumumab to CD38 on the red blood cell surface; however, DTT also inactivates/destroys many antigens on the red blood cell surface by disrupting disulfide bonds. The only antigen system affected that is associated with common, clinically significant antibodies is Kell, making crossmatch testing with K-negative RBCs a reasonable alternative when urgent transfusion is indicated. It is therefore advisable to do a baseline antibody screen and Rh & Kell phenotyping (type and screen) before starting the therapy. If antibody screen is negative, proceed with phenotype matched transfusions during therapy. If antibody screen is positive, give specific antigen negative blood. The incompatibility may persist for up to 6 months after stopping the medicine. Furthermore, blood transfusion centers should be routinely notified when sending such a sample.
Daratumumab can also interfere with flow cytometric evaluation of multiple myeloma, causing an apparent lack of plasma cells.
Recent media
Recent media