Community hub
Decay chain
View on Wikipedia| Nuclear physics |
|---|
In nuclear science a decay chain refers to the predictable series of radioactive disintegrations undergone by the nuclei of certain unstable chemical elements.
Radioactive isotopes do not usually decay directly to stable isotopes, but rather into another radioisotope. The isotope produced by this radioactive emission then decays into another, often radioactive isotope. This chain of decays always terminates in a stable isotope, whose nucleus no longer has the surplus of energy necessary to produce another emission of radiation. Such stable isotopes are then said to have reached their ground states.
The stages or steps in a decay chain are referred to by their relationship to previous or subsequent stages. Hence, a parent isotope is one that undergoes decay to form a daughter isotope. For example element 92, uranium, has an isotope with 144 neutrons (236U) and it decays into an isotope of element 90, thorium, with 142 neutrons (232Th). The daughter isotope may be stable or it may itself decay to form another daughter isotope. 232Th does this when it decays into radium-228. The daughter of a daughter isotope, such as 228Ra, is sometimes called a granddaughter isotope. 228Ra in turn undergoes a further eight decays and transmutations until a stable isotope, 208Pb, is produced, terminating the decay chain of 236U.
The time required for an atom of a parent isotope to decay into its daughter is fundamentally unpredictable and varies widely. For individual nuclei the process is not known to have determinable causes and the time at which it occurs is therefore completely random. The only prediction that can be made is statistical and expresses an average rate of decay. This rate can be represented by adjusting the curve of a decaying exponential distribution with a decay constant (λ) particular to the isotope. On this understanding the radioactive decay of an initial population of unstable atoms over time t follows the curve given by e−λt.
One of the most important properties of any radioactive material follows from this analysis, its half-life. This refers to the time required for half of a given number of radioactive atoms to decay and is inversely related to the isotope's decay constant, λ. Half-lives have been determined in laboratories for many radionuclides, and can range from nearly instantaneous—hydrogen-5 decays in less time than it takes for a photon to go from one end of its nucleus to the other—to fourteen orders of magnitude longer than the age of the universe: tellurium-128 has a half-life of 2.2×1024 years.

The Bateman equation predicts the relative quantities of all the isotopes that compose a given decay chain once that decay chain has proceeded long enough for some of its daughter products to have reached the stable (i.e., nonradioactive) end of the chain. A decay chain that has reached this state, which may require billions of years, is said to be in equilibrium. A sample of radioactive material in equilibrium produces a steady and steadily decreasing quantity of radioactivity as the isotopes that compose it traverse the decay chain. On the other hand, if a sample of radioactive material has been isotopically enriched, meaning that a radioisotope is present in larger quantities than would exist if a decay chain were the only cause of its presence, that sample is said to be out of equilibrium. An unintuitive consequence of this disequilibrium is that a sample of enriched material may occasionally increase in radioactivity as daughter products that are more highly radioactive than their parents accumulate. Both enriched and depleted uranium provide examples of this phenomenon.
History
[edit]The chemical elements came into being in two phases. The first commenced shortly after the Big Bang. From ten seconds to 20 minutes after the beginning of the universe the earliest condensation of light atoms was responsible for the manufacture of the four lightest elements. The vast majority of this primordial production consisted of the three lightest isotopes of hydrogen—protium, deuterium and tritium—and two of the nine known isotopes of helium—helium-3 and helium-4. Trace amounts of lithium-7 and beryllium-7 were likely also produced.
So far as is known, all heavier elements came into being starting around 100 million years later, in a second phase of nucleosynthesis that commenced with the birth of the first stars.[1] The nuclear furnaces that power stellar evolution were necessary to create large quantities of all elements heavier than helium, and the r- and s-processes of neutron capture that occur in stellar cores are thought to have created all such elements up to iron and nickel (atomic numbers 26 and 28). The extreme conditions that attend supernovae explosions are capable of creating the elements between oxygen and rubidium (i.e., atomic numbers 8 through 37). The creation of heavier elements, including those without stable isotopes—all elements with atomic numbers greater than lead's, 82—appears to rely on r-process nucleosynthesis operating amid the immense concentrations of free neutrons released during neutron star mergers.
Most of the isotopes of each chemical element present in the Earth today were formed by such processes no later than the time of our planet's condensation from the solar protoplanetary disc, around 4.5 billion years ago. The exceptions to these so-called primordial elements are those that have resulted from the radioactive disintegration of unstable parent nuclei as they progress down one of several decay chains, each of which terminates with the production of one of the 251 stable isotopes known to exist. Aside from cosmic or stellar nucleosynthesis, and decay chains the only other ways of producing a chemical element rely on atomic weapons, nuclear reactors (natural or manmade) or the laborious atom-by-atom assembly of nuclei with particle accelerators.
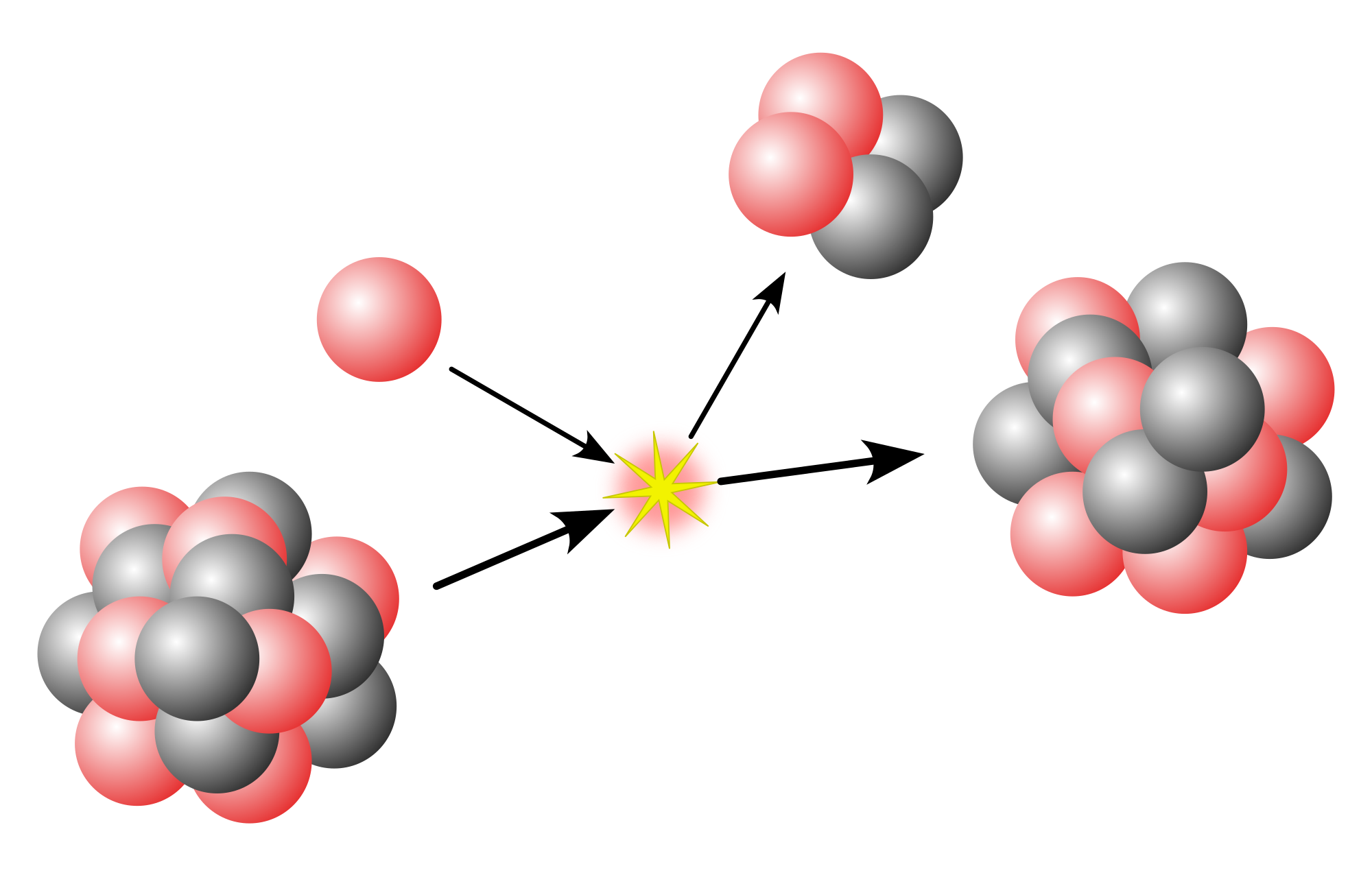
Unstable isotopes decay to their daughter products (which may sometimes be even more unstable) at a given rate; eventually, often after a series of decays, a stable isotope is reached: there are 251 stable isotopes in the universe. In stable isotopes, light elements typically have a lower ratio of neutrons to protons in their nucleus than heavier elements. Light elements such as helium-4 have close to a 1:1 neutron:proton ratio. The heaviest elements such as uranium have close to 1.5 neutrons per proton (e.g. 1.587 in uranium-238). No nuclide heavier than lead-208 is stable; these heavier elements have to shed mass to achieve stability, mostly by alpha decay. The other common way for isotopes with a high neutron to proton ratio (n/p) to decay is beta decay, in which the nuclide changes elemental identity while keeping the same mass number and lowering its n/p ratio. For some isotopes with a relatively low n/p ratio, there is an inverse beta decay, by which a proton is transformed into a neutron, thus moving towards a stable isotope; however, since fission almost always produces products which are neutron heavy, positron emission or electron capture are rare compared to electron emission. There are many relatively short beta decay chains, at least two (a heavy, beta decay and a light, positron decay) for every discrete weight up to around 207 and some beyond, but for the higher mass elements (isotopes heavier than lead) there are only four pathways which encompass all decay chains.[citation needed] This is because there are just two main decay methods: alpha radiation, which reduces the mass number by 4, and beta, which leaves it unchanged. The four paths are termed 4n, 4n + 1, 4n + 2, and 4n + 3; the remainder from dividing the atomic mass by four gives the chain the isotope will follow in its decay. There are other decay modes, but they invariably occur at a lower probability than alpha or beta decay. (It should not be supposed that these chains have no branches: the diagram below shows a few branches of chains, and in reality there are many more, because there are many more isotopes possible than are shown in the diagram.) For example, the third atom of nihonium-278 synthesised underwent six alpha decays down to mendelevium-254, followed by an electron capture (a form of beta decay) to fermium-254, and then a seventh alpha to californium-250,[2] upon which it would have followed the 4n + 2 chain (radium series) as given in this article. However, the heaviest superheavy nuclides synthesised do not reach the four decay chains, because they reach a spontaneously fissioning nuclide after a few alpha decays that terminates the chain: this is what happened to the first two atoms of nihonium-278 synthesised,[3][4] as well as to all heavier nuclides produced.
Three of those chains have a long-lived isotope (or nuclide) near the top; this long-lived nuclide is a bottleneck in the process through which the chain flows very slowly, and keeps the chain below them "alive" with flow. The three long-lived nuclides are uranium-238 (half-life 4.463 billion years), uranium-235 (half-life 704 million years) and thorium-232 (half-life 14.1 billion years). The fourth chain has no such long-lasting bottleneck nuclide near the top, so that chain has long since decayed down to the last before the end: bismuth-209. This nuclide was long thought to be stable, but in 2003 it was found to be unstable, with a very long half-life of 20.1 billion billion years;[5] it is the last step in the chain before stable thallium-205. Because this bottleneck is so long-lived, very small quantities of the final decay product have been produced, and for most practical purposes bismuth-209 is the final decay product.
In the past, during the first few million years of the history of the Solar System, there were more unstable high-mass nuclides in existence, and the four chains were longer, as they included nuclides that have since decayed away. Notably, 244Pu, 237Np, and 247Cm have half-lives over a million years and would have then been bottlenecks higher in the 4n, 4n+1, and 4n+3 chains respectively[6] - 244Pu and 247Cm have been identified as having been present. (There is no nuclide with a half-life over a million years above 238U in the 4n+2 chain.) Today some of these formerly extinct isotopes are again in existence as they have been manufactured. Thus they again take their places in the chain: plutonium-239, used in nuclear weapons, is the major example, decaying to uranium-235 via alpha emission with a half-life 24,500 years. There has also been large-scale production of neptunium-237, resurrecting the extinct fourth chain.[7] The tables below hence start the four decay chains at isotopes of californium with mass numbers from 249 to 252.
| Name of series | Thorium | Neptunium | Uranium | Actinium |
| Mass numbers | 4n | 4n+1 | 4n+2 | 4n+3 |
| Long-lived nuclide | 232Th (244Pu) |
209Bi (237Np) |
238U |
235U (247Cm) |
| Half-life (billions of years) |
14.1 (0.0813) |
20100000000 (0.002144) |
4.463 |
0.704 (0.0156) |
| End of chain | 208Pb | 205Tl | 206Pb | 207Pb |
These four chains are summarised in the chart in the following section.
Types of decay
[edit]
The four most common modes of radioactive decay are: alpha decay, beta decay, inverse beta decay (considered as both positron emission and electron capture), and isomeric transition. Of these decay processes, only alpha decay (fission of a helium-4 nucleus) changes the atomic mass number (A) of the nucleus, and always decreases it by four. Because of this, almost any decay will result in a nucleus whose atomic mass number has the same residue mod 4. This divides the list of nuclides into four classes, each of which forms a main decay chain.
Three of these are readily observed in nature, commonly called the thorium series, the radium or uranium series, and the actinium series, representing three of these four classes, and ending in three different, stable isotopes of lead. The mass number of every isotope in the chain can be represented as A = 4n, A = 4n + 2, or A = 4n + 3, respectively. The long-lived starting isotopes of these three isotopes, respectively thorium-232, uranium-238, and uranium-235, have existed since the formation of the Earth, ignoring the artificial isotopes and their decays created since the 1940s.
Due to the relatively short half-life of its starting isotope neptunium-237 (2.144 million years), the fourth chain, the neptunium series with A = 4n + 1, is already extinct in nature, except for the final rate-limiting step, decay of bismuth-209. Traces of 237Np and its decay products do occur in nature, however, as a result of neutron reactions in uranium ore; neutron capture by natural thorium to give 233U is also possible.[8] The ending isotope of this chain is now known to be thallium-205. Some older sources give the final isotope as bismuth-209, but in 2003 it was discovered that it is very slightly radioactive, with a half-life of 2.01×1019 years.[9]
There are also non-transuranic decay chains of unstable isotopes of light elements, for example those of magnesium-28 and chlorine-39. On Earth, most of the starting isotopes of these chains before 1945 were generated by cosmic radiation. Since 1945, the testing and use of nuclear weapons has also released numerous radioactive fission products. Almost all such isotopes decay by either β− or β+ decay modes, changing from one element to another at the same atomic mass. The later daughter products in such a chain, being closer to beta-stability, generally have the longer half-lives.
Heavy nuclei (actinide) decay chains
[edit]| Actinides[10] by decay chain | Half-life range (a) |
Fission products of 235U by yield[11] | ||||||
|---|---|---|---|---|---|---|---|---|
| 4n (Thorium) |
4n + 1 (Neptunium) |
4n + 2 (Radium) |
4n + 3 (Actinium) |
4.5–7% | 0.04–1.25% | <0.001% | ||
| 228Ra№ | 4–6 a | 155Euþ | ||||||
| 248Bk[12] | > 9 a | |||||||
| 244Cmƒ | 241Puƒ | 250Cf | 227Ac№ | 10–29 a | 90Sr | 85Kr | 113mCdþ | |
| 232Uƒ | 238Puƒ | 243Cmƒ | 29–97 a | 137Cs | 151Smþ | 121mSn | ||
| 249Cfƒ | 242mAmƒ | 141–351 a |
No fission products have a half-life | |||||
| 241Amƒ | 251Cfƒ[13] | 430–900 a | ||||||
| 226Ra№ | 247Bk | 1.3–1.6 ka | ||||||
| 240Pu | 229Th | 246Cmƒ | 243Amƒ | 4.7–7.4 ka | ||||
| 245Cmƒ | 250Cm | 8.3–8.5 ka | ||||||
| 239Puƒ | 24.1 ka | |||||||
| 230Th№ | 231Pa№ | 32–76 ka | ||||||
| 236Npƒ | 233Uƒ | 234U№ | 150–250 ka | 99Tc₡ | 126Sn | |||
| 248Cm | 242Pu | 327–375 ka | 79Se₡ | |||||
| 1.33 Ma | 135Cs₡ | |||||||
| 237Npƒ | 1.61–6.5 Ma | 93Zr | 107Pd | |||||
| 236U | 247Cmƒ | 15–24 Ma | 129I₡ | |||||
| 244Pu | 80 Ma |
... nor beyond 15.7 Ma[14] | ||||||
| 232Th№ | 238U№ | 235Uƒ№ | 0.7–14.1 Ga | |||||
| ||||||||
In the four tables below, very minor branches of decay (branching probability less than one in a million) are omitted. Spontaneous fission is also omitted, though larger than this for the heaviest even nuclei and detectable down to thorium. All nuclear data is taken from [9] unless otherwise noted. The historical names of isotopes are recorded in.[15]
The energy release includes the total kinetic energy of all the emitted particles (electrons, alpha particles, gamma quanta, neutrinos, Auger electrons and X-rays) and the recoiling decay product nucleus; this corresponds to that calculated from atomic masses. The letter 'a' represents a year (from the Latin annus).
In the tables (except for the neptunium series), the historical names of the naturally occurring nuclides are also given. Such names were used at the time when the decay chains were first discovered and investigated; the system listed was only finalized in the 1920s but it would be too confusing to give earlier names also. From these historical names one can thus find the modern isotopic designation.
The three primordial chains given below—thorium, uranium/radium (from uranium-238), and actinium (from uranium-235)—each ends with its own specific lead isotope (lead-208, lead-206, and lead-207 respectively). All the lead isotopes are stable and are also present in nature as primordial nuclides, so their excess amounts in comparison with lead-204 (which has only a primordial origin) are required for accurate uranium–lead dating of rocks. Correlating more than one results in lead-lead dating, capable of even greater accuracy.
Thorium series
[edit]
The 4n chain of thorium-232 is commonly called the "thorium series" or "thorium cascade". The series terminates with lead-208, 6 alpha decays and 4 beta decays from thorium.
Plutonium-244 (which appears several steps above thorium-232) was present in the early Solar System,[6] and is just long-lived enough that it should still survive in trace quantities today,[16] though it probably has not been detected.[17]
The total energy released from thorium-232 to lead-208, including the energy lost to neutrinos, is 42.65 MeV; from californium-252, 71.11 MeV. That last is the largest of the four chains, unsurprisingly for the shell-stability of the product.
| Nuclide | Historic names | Decay mode | Half-life (a = years) |
Energy released MeV |
Decay product | |
|---|---|---|---|---|---|---|
| Short | Long | |||||
| 252Cf | α | 2.645 a | 6.217 | 248Cm | ||
| 248Cm | α | 3.48×105 a | 5.162 | 244Pu | ||
| 244Pu | α | 8.13×107 a | 4.666 | 240U | ||
| 240U | β− | 14.1 h | 0.382 | 240mNp[18] | ||
| 240mNp | IT 0.12% β− 99.88% |
7.22 min | 0.018 2.209 |
240Np 240Pu | ||
| 240Np | β− | 61.9 min | 2.191 | 240Pu | ||
| 240Pu | α | 6561 a | 5.256 | 236U | ||
| 236U | Thoruranium[19] | α | 2.342×107 a | 4.573 | 232Th | |
| 232Th | Th | Thorium | α | 1.40×1010 a | 4.082 | 228Ra |
| 228Ra | MsTh1 | Mesothorium 1 | β− | 5.75 a | 0.046 | 228Ac |
| 228Ac | MsTh2 | Mesothorium 2 | β− | 6.15 h | 2.123 | 228Th |
| 228Th | RdTh | Radiothorium | α | 1.9125 a | 5.520 | 224Ra |
| 224Ra | ThX | Thorium X | α | 3.632 d | 5.789 | 220Rn |
| 220Rn | Tn | Thoron, Thorium Emanation |
α | 55.6 s | 6.405 | 216Po |
| 216Po | ThA | Thorium A | α | 0.144 s | 6.906 | 212Pb |
| 212Pb | ThB | Thorium B | β− | 10.627 h | 0.569 | 212Bi |
| 212Bi | ThC | Thorium C | β− 64.06% α 35.94% |
60.55 min | 2.252 6.207 |
212Po 208Tl |
| 212Po | ThC′ | Thorium C′ | α | 294.4 ns | 8.954 | 208Pb |
| 208Tl | ThC″ | Thorium C″ | β− | 3.053 min | 4.999 | 208Pb |
| 208Pb | ThD | Thorium D | stable | |||
Neptunium series
[edit].svg)
The 4n+1 chain of neptunium-237 is commonly called the "neptunium series" or "neptunium cascade". In this series, only two of the isotopes involved are found naturally in significant quantities, namely the final two: bismuth-209 and thallium-205. Some of the other isotopes have been detected in nature, originating from trace quantities of 237Np produced by the (n,2n) knockout reaction in primordial 238U.[8]
Since this series was only discovered and studied in 1947–1948,[20] its nuclides were never given historic names. Uniquely among the four, this decay chain has an isotope of radon only produced in a rare branch (not shown in the illustration) but not in the main decay sequence; thus, radon from this decay chain will hardly migrate through rock. Also uniquely, it ends in thallium (or, practically speaking, bismuth) rather than lead. This series terminates with the stable isotope thallium-205, 8 alpha decays and 4 beta decays from neptunium.
The total energy released from neptunium-237 to thallium-205, including the energy lost to neutrinos, is 49.29 MeV; from californium-249, 66.87 MeV. As the energy of the final step from bismuth to thallium, though known, will not be available until the inconceivable future, it may be better to quote the figures 46.16 MeV and 63.73 MeV to bismuth-209.
| Nuclide | Decay mode | Half-life (a = years) |
Energy released MeV |
Decay product |
|---|---|---|---|---|
| 249Cf | α | 351 a | 6.293 | 245Cm |
| 245Cm | α | 8250 a | 5.624 | 241Pu |
| 241Pu | β− 99.9975% α 0.0025% |
14.33 a | 0.021 5.140 |
241Am 237U |
| 241Am | α | 432.6 a | 5.638 | 237Np |
| 237U | β− | 6.752 d | 0.518 | 237Np |
| 237Np | α | 2.144×106 a | 4.957 | 233Pa |
| 233Pa | β− | 26.98 d | 0.570 | 233U |
| 233U | α | 1.592×105 a | 4.909 | 229Th |
| 229Th | α | 7920 a | 5.168 | 225Ra |
| 225Ra | β− 99.9974% α 0.0026%[21][a] |
14.8 d | 0.356 5.097 |
225Ac 221Rn |
| 225Ac | α | 9.919 d | 5.935 | 221Fr |
| 221Rn | β− 78% α 22% |
25.7 min | 1.194 6.163 |
221Fr 217Po |
| 221Fr | α 99.9952% β− 0.0048% |
4.801 min | 6.457 0.313 |
217At 221Ra |
| 221Ra | α | 25 s | 6.880 | 217Rn |
| 217Po | α 97.5% β− 2.5% |
1.53 s | 6.662 1.488 |
213Pb 217At |
| 217At | α 99.992% β− 0.008% |
32.6 ms | 7.202 0.736 |
213Bi 217Rn |
| 217Rn | α | 590 μs | 7.888 | 213Po |
| 213Pb | β− | 10.2 min | 2.028 | 213Bi |
| 213Bi | β− 97.91% α 2.09% |
45.6 min | 1.422 5.988 |
213Po 209Tl |
| 213Po | α | 3.705 μs | 8.536 | 209Pb |
| 209Tl | β− | 2.162 min | 3.970 | 209Pb |
| 209Pb | β− | 3.235 h | 0.644 | 209Bi |
| 209Bi | α | 2.01×1019 a | 3.137 | 205Tl |
| 205Tl | stable |
- ^ The value .026%, found at other places, is a typographical error. The original data is cited here.
Uranium series
[edit].svg)
{kind=link}
The 4n+2 chain of uranium-238 is called the "uranium series" or "radium series", the latter from the first member known when it was named, radium-226. The series terminates with lead-206, 8 alpha decays and 6 beta decays from uranium.
The total energy released from uranium-238 to lead-206, including the energy lost to neutrinos, is 51.69 MeV; from californium-250, 68.28 MeV.
| Nuclide | Historic names | Decay mode | Half-life (a = years) |
Energy released MeV |
Decay product | |
|---|---|---|---|---|---|---|
| Short | Long | |||||
| 250Cf | α | 13.08 a | 6.128 | 246Cm | ||
| 246Cm | α | 4760 a | 5.475 | 242Pu | ||
| 242Pu | α | 3.75×105 a | 4.984 | 238U | ||
| 238U | UI | Uranium I | α | 4.463×109 a | 4.270 | 234Th |
| 234Th | UX1 | Uranium X1 | β− | 24.11 d | 0.195 | 234mPa[18] |
| 234mPa | UX2, Bv | Uranium X2 Brevium |
IT 0.16% β− 99.84% |
1.16 min | 0.079 2.273 |
234Pa 234U |
| 234Pa | UZ | Uranium Z | β− | 6.70 h | 2.194 | 234U |
| 234U | UII | Uranium II | α | 2.455×105 a | 4.858 | 230Th |
| 230Th | Io | Ionium | α | 7.54×104 a | 4.770 | 226Ra |
| 226Ra | Ra | Radium | α | 1600 a | 4.871 | 222Rn |
| 222Rn | Rn | Radon, Radium Emanation |
α | 3.8215 d | 5.590 | 218Po |
| 218Po | RaA | Radium A | α 99.98% β− 0.02% |
3.097 min | 6.115 0.257 |
214Pb 218At |
| 218At | α 100% β− |
1.28 s | 6.876 2.883 |
214Bi 218Rn | ||
| 218Rn | α | 33.75 ms | 7.262 | 214Po | ||
| 214Pb | RaB | Radium B | β− | 27.06 min | 1.018 | 214Bi |
| 214Bi | RaC | Radium C | β− 99.979% α 0.021% |
19.9 min | 3.269 5.621 |
214Po 210Tl |
| 214Po | RaC' | Radium C' | α | 163.5 μs | 7.833 | 210Pb |
| 210Tl | RaC" | Radium C" | β− β−n 0.009% |
1.30 min | 5.481 0.296 |
210Pb 209Pb (in neptunium series) |
| 210Pb | RaD | Radium D | β− α 1.9×10−6% |
22.2 a | 0.0635 3.793 |
210Bi 206Hg |
| 210Bi | RaE | Radium E | β− α 1.32×10−4% |
5.012 d | 1.161 5.035 |
210Po 206Tl |
| 210Po | RaF | Radium F | α | 138.376 d | 5.407 | 206Pb |
| 206Hg | β− | 8.32 min | 1.307 | 206Tl | ||
| 206Tl | RaE" | Radium E" | β− | 4.20 min | 1.532 | 206Pb |
| 206Pb | RaG | Radium G | stable | |||
Actinium series
[edit]
{kind=link}
The 4n+3 chain of uranium-235 is commonly called the "actinium series" or "actinium cascade", from the first member known when it was named, actinium-227. This series terminates with lead-207, 7 alpha decays and 4 beta decays from uranium.
In the early Solar System, this chain went back to 247Cm. This manifests itself today as variations in 235U/238U ratios, since curium and uranium have noticeably different chemistries and therefore partitioned differently.[6][22]
The total energy released from uranium-235 to lead-207, including the energy lost to neutrinos, is 46.40 MeV; from californium-251, 69.91 MeV.
| Nuclide | Historic name | Decay mode | Half-life (a = years) |
Energy released MeV |
Decay product | |
|---|---|---|---|---|---|---|
| Short | Long | |||||
| 251Cf | α | 900 a | 6.177 | 247Cm | ||
| 247Cm | α | 1.56×107 a | 5.353 | 243Pu | ||
| 243Pu | β− | 4.955 h | 0.578 | 243Am | ||
| 243Am | α | 7350 a | 5.439 | 239Np | ||
| 239Np | β- | 2.356 d | 0.723 | 239Pu | ||
| 239Pu | α | 2.411×104 a | 5.244 | 235U | ||
| 235U | AcU | Actino-uranium | α | 7.04×108 a | 4.678 | 231Th |
| 231Th | UY | Uranium Y | β− | 25.52 h | 0.391 | 231Pa |
| 231Pa | Pa | Protoactinium | α | 3.27×104 a | 5.150 | 227Ac |
| 227Ac | Ac | Actinium | β− 98.62% α 1.38% |
21.772 a | 0.045 5.042 |
227Th 223Fr |
| 227Th | RdAc | Radioactinium | α | 18.693 d | 6.147 | 223Ra |
| 223Fr | AcK | Actinium K | β− 99.994% α 0.006% |
22.00 min | 1.149 5.561 |
223Ra 219At |
| 223Ra | AcX | Actinium X | α | 11.435 d | 5.979 | 219Rn |
| 219At | α 93.6% β− 6.4% |
56 s | 6.342 1.567 |
215Bi 219Rn | ||
| 219Rn | An | Actinon, Actinium Emanation |
α | 3.96 s | 6.946 | 215Po |
| 215Bi | β− | 7.6 min | 2.171 | 215Po | ||
| 215Po | AcA | Actinium A | α β− 2.3×10−4% |
1.781 ms | 7.526 0.715 |
211Pb 215At |
| 215At | α | 37 μs | 8.177 | 211Bi | ||
| 211Pb | AcB | Actinium B | β− | 36.16 min | 1.366 | 211Bi |
| 211Bi | AcC | Actinium C | α 99.724% β− 0.276% |
2.14 min | 6.750 0.573 |
207Tl 211Po |
| 211Po | AcC' | Actinium C' | α | 516 ms | 7.595 | 207Pb |
| 207Tl | AcC" | Actinium C" | β− | 4.77 min | 1.418 | 207Pb |
| 207Pb | AcD | Actinium D | stable | |||
See also
[edit]Notes
[edit]- ^ Bromm, Richard B. Larson, Volker. "The First Stars in the Universe". Scientific American. Retrieved 2024-09-29.
{{cite web}}: CS1 maint: multiple names: authors list (link) - ^ K. Morita; Morimoto, Kouji; Kaji, Daiya; Haba, Hiromitsu; Ozeki, Kazutaka; Kudou, Yuki; Sumita, Takayuki; Wakabayashi, Yasuo; Yoneda, Akira; Tanaka, Kengo; et al. (2012). "New Results in the Production and Decay of an Isotope, 278113, of the 113th Element". Journal of the Physical Society of Japan. 81 (10) 103201. arXiv:1209.6431. Bibcode:2012JPSJ...81j3201M. doi:10.1143/JPSJ.81.103201. S2CID 119217928.
- ^ Morita, Kosuke; Morimoto, Kouji; Kaji, Daiya; Akiyama, Takahiro; Goto, Sin-Ichi; Haba, Hiromitsu; Ideguchi, Eiji; Kanungo, Rituparna; et al. (2004). "Experiment on the Synthesis of Element 113 in the Reaction 209Bi(70Zn, n)278113". Journal of the Physical Society of Japan. 73 (10): 2593–2596. Bibcode:2004JPSJ...73.2593M. doi:10.1143/JPSJ.73.2593.
- ^ Barber, Robert C.; Karol, Paul J; Nakahara, Hiromichi; Vardaci, Emanuele; Vogt, Erich W. (2011). "Discovery of the elements with atomic numbers greater than or equal to 113 (IUPAC Technical Report)". Pure and Applied Chemistry. 83 (7): 1485. doi:10.1351/PAC-REP-10-05-01.
- ^ J.W. Beeman; et al. (2012). "First Measurement of the Partial Widths of 209Bi Decay to the Ground and to the First Excited States". Physical Review Letters. 108 (6) 062501. arXiv:1110.3138. doi:10.1103/PhysRevLett.108.062501. PMID 22401058. S2CID 118686992.
- ^ a b c Davis, Andrew M. (2022). "Short-Lived Nuclides in the Early Solar System: Abundances, Origins, and Applications". Annual Review of Nuclear and Particle Science. 72: 339–363. Bibcode:2022ARNPS..72..339D. doi:10.1146/annurev-nucl-010722-074615.
- ^ Koch, Lothar (2000). Transuranium Elements, in Ullmann's Encyclopedia of Industrial Chemistry. Wiley. doi:10.1002/14356007.a27_167.
- ^ a b Peppard, D. F.; Mason, G. W.; Gray, P. R.; Mech, J. F. (1952). "Occurrence of the (4n + 1) series in nature" (PDF). Journal of the American Chemical Society. 74 (23): 6081–6084. Bibcode:1952JAChS..74.6081P. doi:10.1021/ja01143a074.
- ^ a b Kondev, F. G.; Wang, M.; Huang, W. J.; Naimi, S.; Audi, G. (2021). "The NUBASE2020 evaluation of nuclear properties" (PDF). Chinese Physics C. 45 (3) 030001. doi:10.1088/1674-1137/abddae.
- ^ Plus radium (element 88). While actually a sub-actinide, it immediately precedes actinium (89) and follows a three-element gap of instability after polonium (84) where no nuclides have half-lives of at least four years (the longest-lived nuclide in the gap is radon-222 with a half life of less than four days). Radium's longest lived isotope, at 1,600 years, thus merits the element's inclusion here.
- ^ Specifically from thermal neutron fission of uranium-235, e.g. in a typical nuclear reactor.
- ^ Milsted, J.; Friedman, A. M.; Stevens, C. M. (1965). "The alpha half-life of berkelium-247; a new long-lived isomer of berkelium-248". Nuclear Physics. 71 (2): 299. Bibcode:1965NucPh..71..299M. doi:10.1016/0029-5582(65)90719-4.
"The isotopic analyses disclosed a species of mass 248 in constant abundance in three samples analysed over a period of about 10 months. This was ascribed to an isomer of Bk248 with a half-life greater than 9 [years]. No growth of Cf248 was detected, and a lower limit for the β− half-life can be set at about 104 [years]. No alpha activity attributable to the new isomer has been detected; the alpha half-life is probably greater than 300 [years]." - ^ This is the heaviest nuclide with a half-life of at least four years before the "sea of instability".
- ^ Excluding those "classically stable" nuclides with half-lives significantly in excess of 232Th; e.g., while 113mCd has a half-life of only fourteen years, that of 113Cd is eight quadrillion years.
- ^ Thoennessen, M. (2016). The Discovery of Isotopes: A Complete Compilation. Springer. p. 19. doi:10.1007/978-3-319-31763-2. ISBN 978-3-319-31761-8. LCCN 2016935977.
- ^ Hoffman, D. C.; Lawrence, F. O.; Mewherter, J. L.; Rourke, F. M. (1971). "Detection of Plutonium-244 in Nature". Nature. 234 (5325): 132–134. Bibcode:1971Natur.234..132H. doi:10.1038/234132a0. S2CID 4283169.
- ^ Lachner, J.; et al. (2012). "Attempt to detect primordial 244Pu on Earth". Physical Review C. 85 (1) 015801. Bibcode:2012PhRvC..85a5801L. doi:10.1103/PhysRevC.85.015801.
- ^ a b ENSDF analysis available at National Nuclear Data Center. "NuDat 3.0 database". Brookhaven National Laboratory.
- ^ Trenn, Thaddeus J. (1978). "Thoruranium (U-236) as the extinct natural parent of thorium: The premature falsification of an essentially correct theory". Annals of Science. 35 (6): 581–97. doi:10.1080/00033797800200441.
- ^ Thoennessen, M. (2016). The Discovery of Isotopes: A Complete Compilation. Springer. p. 20. doi:10.1007/978-3-319-31763-2. ISBN 978-3-319-31761-8. LCCN 2016935977.
- ^ Liang, C. F.; Paris, P.; Sheline, R. K. (2000-09-19). "α decay of 225Ra". Physical Review C. 62 (4) 047303. American Physical Society (APS). Bibcode:2000PhRvC..62d7303L. doi:10.1103/physrevc.62.047303. ISSN 0556-2813.
- ^ Tsaletka, R.; Lapitskii, A. V. (1960). "Occurrence of the Transuranium Elements in Nature". Russian Chemical Reviews. 29 (12): 684–689. Bibcode:1960RuCRv..29..684T. doi:10.1070/RC1960v029n12ABEH001264. Retrieved 20 January 2024.
References
[edit]- C.M. Lederer; J.M. Hollander; I. Perlman (1968). Table of Isotopes (6th ed.). New York: John Wiley & Sons.
External links
[edit]- Nucleonica nuclear science portal
- Nucleonica's Decay Engine for professional online decay calculations
- EPA – Radioactive Decay
- Government website listing isotopes and decay energies
- National Nuclear Data Center – freely available databases that can be used to check or construct decay chains
 IAEA – Live Chart of Nuclides (with decay chains)
IAEA – Live Chart of Nuclides (with decay chains)- Decay Chain Finder
Decay chain
View on GrokipediaFundamentals
Definition and Overview
A decay chain, also known as a radioactive decay series, is a sequence of radioactive decays in which an unstable atomic nucleus (nuclide) undergoes successive transformations into other nuclides, either stable or unstable, by emitting ionizing particles or radiation, until a stable end product is ultimately reached.[4] This process begins with a long-lived parent nuclide and proceeds through intermediate daughter nuclides, each of which may itself be radioactive.[5] The general structure of a decay chain can be linear, where each step leads to a single successor, or branched, where a given nuclide can decay via multiple pathways, resulting in parallel sequences that reconverge toward stability.[6] These chains typically culminate in stable isotopes, most commonly the lead isotopes ^{206}Pb, ^{207}Pb, or ^{208}Pb, depending on the parent nuclide involved.[7] For example, a simplified schematic of a decay chain might appear as follows:Key Concepts in Radioactive Decay
Radioactive decay follows an exponential law, where the number of undecayed nuclei $ N(t) $ at time $ t $ is given by $ N(t) = N_0 e^{-\lambda t} $, with $ \lambda $ being the decay constant representing the probability of decay per unit time for a single nucleus.[9] The decay constant $ \lambda $ has units of inverse time, typically s, and quantifies the intrinsic instability of the radionuclide.[9] The half-life $ t_{1/2} $ is the time required for half of the radioactive atoms in a sample to decay, providing a practical measure of decay rate independent of the initial number of atoms. It relates to the decay constant by the formula $ t_{1/2} = \frac{\ln 2}{\lambda} \approx \frac{0.693}{\lambda} $, ensuring that after each half-life interval, the remaining activity halves.[10] The activity $ A $ of a radioactive sample, defined as the rate of decay (disintegrations per unit time), is expressed as $ A = \lambda N $, where $ N $ is the number of radioactive atoms present.[11] In decay chains, activity levels evolve based on this relation for each nuclide. Secular equilibrium arises in a parent-daughter pair when the parent's half-life greatly exceeds the daughter's ($ t_{1/2,\text{parent}} \gg t_{1/2,\text{daughter}} $), causing the daughter's activity to approach equality with the parent's after sufficient time, as production matches decay.[12] This condition simplifies analysis of long-lived parents supporting short-lived progeny in natural series. For multi-step decay chains, the Bateman equations provide the general analytical solution for the number of atoms of the $ i $-th nuclide at time $ t $, expressed as $ N_i(t) = \sum $ terms involving exponential factors with decay constants $ \lambda_j $ for $ j = 1 $ to $ i $, weighted by initial abundances and differences $ \lambda_j - \lambda_i $.[13] These equations, derived in 1910, account for successive transformations without assuming equilibrium.[14] Activity is measured in becquerels (Bq), the SI unit defined as one decay per second.[15] The historical curie (Ci) equals $ 3.7 \times 10^{10} $ Bq, originally based on the activity of 1 gram of radium-226.[15]Historical Development
Early Discoveries
The discovery of radioactivity began with Henri Becquerel's accidental observation in 1896, when he found that uranium salts emitted penetrating rays capable of exposing photographic plates even in the absence of light, initially mistaken for phosphorescence but soon recognized as a spontaneous emission independent of external excitation.[16] This finding, termed "uranic rays," marked the first evidence of natural radioactivity and prompted investigations into similar emissions, or "emanations," from uranium compounds.[17] Building on Becquerel's work, Marie and Pierre Curie isolated two highly radioactive elements from pitchblende ore in 1898: polonium, named after Marie's native Poland, and radium, derived from the Latin for "ray," both exhibiting far greater activity than uranium.[18] Their research also contributed to the classification of the emissions from these substances into three types—alpha rays (heavily ionizing and least penetrating), beta rays (deflected by magnetic fields like electrons), and gamma rays (highly penetrating electromagnetic radiation)—laying the groundwork for understanding the diverse manifestations of radioactive decay.[18] These discoveries demonstrated that radioactivity was an atomic property inherent to certain elements, not merely a secondary effect.[19] In 1900, German physicist Friedrich Ernst Dorn identified a radioactive gas as an intermediate product in the decay of radium, which was part of the uranium decay sequence, initially calling it "radium emanation" due to its gaseous nature and ability to diffuse from solid sources.[20] This emanation, later recognized as radon, provided early evidence of gaseous intermediates in radioactive transformations, bridging the gap between parent elements and their decay products.[17] Ernest Rutherford and Frederick Soddy advanced the field significantly between 1902 and 1903 through experiments on thorium compounds, where they separated a highly radioactive "thorium X" from thorium oxide and observed its gradual reformation, proving that radioactivity involved the spontaneous transmutation of one element into another.[21] They proposed the concept of "radioactive genealogy," a series of successive transformations where unstable atoms decay into daughter products, each potentially radioactive, challenging the immutability of elements and establishing the framework for decay chains.[21] By the 1910s, systematic studies had identified approximately 30 to 40 radioactive nuclides across emerging decay chains, including key members like radium, radon, and various short-lived intermediates in the uranium and thorium series, through chemical separations and ionization measurements.[17]Modern Understanding and Mapping
In the 1930s and 1940s, advancements in instrumentation such as cloud chambers and mass spectrometry enabled the full mapping of several radioactive decay chains by visualizing particle tracks and identifying isotopic masses with greater precision.[22][23] Cloud chambers, refined during cosmic ray studies, captured alpha and beta particle paths from decay events, while early mass spectrometers separated decay products based on mass-to-charge ratios, confirming sequences in thorium and uranium series.[24] A pivotal contribution came from Lise Meitner and Otto Frisch's 1939 interpretation of nuclear fission, which linked neutron-induced uranium splitting to branched decay chains of fission products, explaining observed beta decay sequences and gamma emissions.[25][26] The Manhattan Project in the 1940s accelerated detailed studies of actinide decay chains, driven by the need to understand plutonium production and fission product behavior for nuclear weapon design.[27] Researchers at Los Alamos and other sites mapped alpha and beta decays in transuranic elements like neptunium-237 and plutonium-239, using cyclotron-produced samples to trace chain progressions and half-lives essential for chain reaction control.[28] These efforts revealed complex branching in actinide series, informing safety protocols and material stability under irradiation.[29] Following the 1950s, decay chain research expanded into geochemical and astrophysical contexts, with the identification of extinct series providing insights into Earth's formation and stellar nucleosynthesis. Geochemists applied uranium-series disequilibria to date ocean sediments and volcanic rocks, leveraging alpha recoil and radon diffusion in chains for timescale resolution up to 500,000 years.[30] In astrophysics, modeling of r-process pathways incorporated decay chains to explain heavy element abundances in neutron star mergers.[31] The neptunium series, originating from now-extinct 237Np (half-life 2.14 million years), was fully outlined in the 1950s through synthesis and decay tracking, revealing its role in primordial actinide inventories.[32] Contemporary mapping relies on alpha spectroscopy, mass spectrometry, and computational modeling to resolve branching ratios and minor pathways. Alpha spectroscopy distinguishes nuclides by emission energies (e.g., 5-9 MeV peaks), enabling chain identification in environmental samples with resolutions below 20 keV.[33] Thermal ionization mass spectrometry provides isotopic abundance data for tracing ingrowth in long-lived parents like 238U.[34] Computational tools, such as Monte Carlo simulations, predict branching fractions by integrating nuclear shell models with decay probabilities, aiding predictions for superheavy elements.[35][36] As of 2025, high-precision experiments at national laboratories continue to refine details of actinide chains and fission product decays. For example, measurements at CERN's ISOLDE facility have determined half-lives for isotopes in natural decay series, such as 215At (36.3 μs) and 221Ra (26.2 s), and improved mass uncertainties for neutron-deficient tin isotopes, common fission products.[37][38]Decay Processes Involved
Alpha and Beta Decay
Alpha decay is a radioactive process in which an unstable atomic nucleus emits an alpha particle, consisting of a helium-4 nucleus (two protons and two neutrons), resulting in a daughter nucleus with atomic mass number reduced by 4 and atomic number decreased by 2.[39] This decay mode is energetically possible when the Q-value, defined as the energy released, is positive:Other Decay Modes
Gamma decay involves the emission of high-energy photons from an excited nucleus, serving primarily to de-excite the nucleus without altering its atomic number (Z) or mass number (A). This process typically follows alpha or beta decay, where the daughter nucleus is left in an excited state, and the gamma emission releases the excess energy to reach a lower energy level. In natural decay chains, such as the uranium and thorium series, gamma rays are prominent from specific nuclides; for instance, in the uranium-238 chain, ^{214}Pb and ^{214}Bi are major gamma emitters, contributing significantly to the radiation profile during the chain's progression. Similarly, in the thorium-232 chain, ^{228}Ac, ^{212}Pb, and ^{208}Tl emit characteristic gamma rays that aid in identifying chain stages through spectroscopy.[47] Internal conversion competes with gamma decay as an alternative de-excitation mechanism, where the nuclear excitation energy is transferred directly to an orbital electron, ejecting it from the atom rather than emitting a photon. This electromagnetic process is more probable for low-energy transitions and higher multipolarities, with the conversion coefficient α indicating the ratio of conversion to gamma emission probabilities, often favoring internal conversion in heavy nuclei due to stronger Coulomb interactions. In decay chains, internal conversion electrons from nuclides like those in actinide series provide additional signatures for tracing chain evolution, though they are less penetrating than gamma rays and thus play a secondary role in energy balance.[48] Electron capture (EC) is a weak interaction process where a proton-rich nucleus captures an inner-shell orbital electron, transforming a proton into a neutron, thereby decreasing Z by 1 while A remains unchanged, and emitting a neutrino. This mode is prevalent in proton-excess heavy nuclides where the energy available is insufficient for positron emission, such as in certain neutron-deficient actinides within synthetic branches of natural chains. In decay contexts, EC contributes to branching pathways in heavy elements, often accompanied by X-ray emission from atomic electron rearrangements, and helps populate excited states that may lead to subsequent gamma or conversion processes.[49] Rare decay modes in decay chains include spontaneous fission (SF), cluster decay, and beta-delayed processes, which occur primarily in heavy actinides and introduce alternative termination or branching points. Spontaneous fission involves the quantum tunneling of a heavy nucleus through its fission barrier, splitting into two fragments and neutrons without external stimulation, terminating chains in elements like uranium-238 (with a partial half-life of ~10^{16} years) and becoming dominant in superheavy actinides. Cluster decay, positioned between alpha decay and SF, entails the emission of a preformed cluster heavier than an alpha particle (e.g., ^{14}C from ^{222}Ra or ^{20}Ne from uranium isotopes), with branching ratios around 10^{-10} to 10^{-13}, offering insights into nuclear structure in transuranic chains. Beta-delayed processes, such as beta-delayed fission (βDF), occur when beta decay populates an excited daughter state above the fission barrier, leading to fission with low probabilities (~3 \times 10^{-5} or less) in neutron-rich precursors like ^{180}Tl, influencing chain dynamics in r-process nucleosynthesis scenarios. These modes, though infrequent, are crucial for understanding stability limits and energy dissipation in long decay sequences of heavy elements.[50][51][52]Natural Decay Series
Thorium Series
The thorium series, designated as the 4n decay chain, originates from the primordial radionuclide thorium-232 (²³²Th), which has a half-life of 1.405 × 10¹⁰ years and decays primarily via alpha emission.[53] This series consists of 11 radioactive nuclides that undergo a total of six alpha decays and four beta-minus decays, culminating in the stable isotope lead-208 (²⁰⁸Pb).[8] The chain is significant in natural radioactivity due to its presence in the Earth's crust and its role in environmental radiation exposure. The decay sequence begins with ²³²Th undergoing alpha decay to radium-228 (²²⁸Ra), followed by beta-minus decay to actinium-228 (²²⁸Ac), and another beta-minus decay to thorium-228 (²²⁸Th). Subsequent alpha decays proceed through radium-224 (²²⁴Ra) and radon-220 (²²⁰Rn, known as thoron with a half-life of 55.6 seconds) to polonium-216 (²¹⁶Po), then beta-minus decay via lead-212 (²¹²Pb) to bismuth-212 (²¹²Bi). At ²¹²Bi, the chain branches: approximately 64% proceeds via beta-minus decay to polonium-212 (²¹²Po), which undergoes alpha decay to ²⁰⁸Pb, while 36% occurs via alpha decay to thallium-208 (²⁰⁸Tl), followed by beta-minus decay to ²⁰⁸Pb.[8] The following table summarizes the nuclides in the thorium-232 decay series, including decay modes and half-lives:| Nuclide | Half-Life | Decay Mode |
|---|---|---|
| ²³²Th | 1.4 × 10¹⁰ years | α |
| ²²⁸Ra | 5.75 years | β⁻ |
| ²²⁸Ac | 6.13 hours | β⁻ |
| ²²⁸Th | 1.91 years | α |
| ²²⁴Ra | 3.66 days | α |
| ²²⁰Rn | 55.6 seconds | α |
| ²¹⁶Po | 0.145 seconds | α |
| ²¹²Pb | 10.64 hours | β⁻ |
| ²¹²Bi | 60.55 minutes | β⁻ (64%), α (36%) |
| ²¹²Po | 0.299 μs | α |
| ²⁰⁸Tl | 3.053 minutes | β⁻ |
| ²⁰⁸Pb | Stable | — |
Uranium-Radium Series
The Uranium-Radium series, also known as the radium series or 4n+2 decay chain, is one of the four natural radioactive decay chains and the most abundant in the Earth's crust due to the prevalence of its parent nuclide. It commences with uranium-238 (²³⁸U), the primary isotope of uranium comprising over 99% of natural uranium deposits, which undergoes alpha decay with a half-life of 4.468 billion years. This extraordinarily long half-life renders ²³⁸U effectively primordial, having persisted since the formation of the solar system. The series proceeds through a sequence of 14 successive decays—eight alpha emissions and six beta-minus decays—culminating in the stable end product lead-206 (²⁰⁶Pb).[56][57]/21%3A_Nuclear_Chemistry/21.03%3A_Radioactive_Decay) The decay pathway is predominantly linear, exhibiting minimal branching and thus predictable accumulation of daughters under equilibrium conditions. Initial steps involve alpha decay of ²³⁸U to thorium-234 (²³⁴Th, half-life 24.1 days), followed by two rapid beta-minus decays via protactinium-234 (²³⁴Pa) to uranium-234 (²³⁴U, half-life 245,500 years), a notable long-lived intermediate that contributes significantly to the series' overall activity. Subsequent alpha decays yield thorium-230 (²³⁰Th, half-life 75,380 years), radium-226 (²²⁶Ra, half-life 1,600 years), and radon-222 (²²²Rn, half-life 3.82 days), the latter being a radioactive noble gas that readily emanates from minerals and poses inhalation risks. The chain continues through shorter-lived polonium, lead, bismuth, and astatine isotopes before terminating at ²⁰⁶Pb.[58] This series is ubiquitous in the continental crust at concentrations of 1–3 parts per million for uranium, as well as in seawater (typically 3–4 micrograms per liter), influencing global geochemical cycles. Its presence forms the foundation for uranium-lead (U-Pb) geochronology, a technique that measures the ratio of ²³⁸U to ²⁰⁶Pb in minerals like zircon to date geological events spanning billions of years.[59]Actinium Series
The actinium series, one of the four natural radioactive decay chains, originates from the primordial isotope uranium-235 (²³⁵U), which constitutes approximately 0.72% of natural uranium deposits.[60] This isotope is fissile, meaning it can sustain a nuclear chain reaction when bombarded by thermal neutrons, making it essential for nuclear fuel cycles in reactors. With a half-life of 704 million years, ²³⁵U undergoes alpha decay to initiate the sequence, proceeding through a relatively short chain compared to other series due to its intermediate longevity among actinides. The decay pathway involves seven alpha decays and four beta-minus decays, reducing the mass number by 28 and the atomic number from 92 to 82, ultimately yielding the stable end product lead-207 (²⁰⁷Pb).[61] Key initial steps include: ²³⁵U decaying via alpha emission to thorium-231 (²³¹Th), which then undergoes beta-minus decay to protactinium-231 (²³¹Pa); ²³¹Pa follows with alpha decay to actinium-227 (²²⁷Ac).[58] Continuing, ²²⁷Ac decays primarily via beta-minus to thorium-227 (²²⁷Th) but exhibits branching with a 1.38% probability of alpha decay directly to francium-223 (²²³Fr), while the remaining 98.62% proceeds through the beta path.[62] From ²²³Fr (via the minor branch) or ²²³Ra (from ²²⁷Th alpha decay), the chain advances to radon-219 (²¹⁹Rn) via alpha decay of ²²³Ra (or beta-minus from ²²³Fr to ²²³Ra then alpha). After ²¹⁹Rn, the main path proceeds via alpha decay to astatine-215 (²¹⁵At), which undergoes alpha decay to bismuth-211 (²¹¹Bi). ²¹¹Bi then decays primarily (~99.7%) via beta-minus to polonium-211 (²¹¹Po), followed by alpha decay to ²⁰⁷Pb; a minor branch (~0.3%) from ²¹¹Bi is alpha decay to thallium-207 (²⁰⁷Tl), followed by beta-minus to ²⁰⁷Pb. A minor branch (~0.8%) from ²¹⁵At involves beta-minus decay to ²¹⁵Po, which beta-minus decays to lead-211 (²¹¹Pb), then beta-minus to ²¹¹Bi, rejoining the main chain.[63] The following table summarizes the principal nuclides in the actinium series (uranium-235 decay chain), focusing on the main pathway with notable branches indicated:| Nuclide | Half-Life | Decay Mode |
|---|---|---|
| ²³⁵U | 7.04 × 10⁸ years | α |
| ²³¹Th | 25.52 hours | β⁻ |
| ²³¹Pa | 3.28 × 10⁴ years | α |
| ²²⁷Ac | 21.77 years | β⁻ (98.62%), α (1.38%) |
| ²²⁷Th | 18.72 days | α |
| ²²³Ra | 11.43 days | α |
| ²¹⁹Rn | 3.96 seconds | α |
| ²¹⁵At | 1.0 × 10⁻⁴ seconds | α (~99.2%), β⁻ (~0.8%) |
| ²¹¹Bi | 2.14 minutes | β⁻ (~99.7%), α (~0.3%) |
| ²¹¹Po | 5.16 × 10⁻¹ seconds | α |
| ²⁰⁷Tl | 4.77 minutes | β⁻ |
| ²⁰⁷Pb | Stable | — |
Neptunium Series
The neptunium series, also known as the 4n+1 radioactive decay series, originates from the artificial isotope neptunium-237 and terminates at the stable bismuth-209. This chain is not primordial and exists primarily as a result of human activities, particularly nuclear reactor operations, where neptunium-237 accumulates as a byproduct without significant natural occurrence due to the rapid decay of potential precursor isotopes in Earth's early history. Neptunium-237, the parent nuclide with a half-life of 2.144 × 10⁶ years, forms mainly through two pathways: the beta decay of uranium-237 (half-life 6.75 days), itself produced via the (n,2n) reaction on uranium-238 in reactor fuel, or the alpha decay of americium-241 (half-life 432.2 years), a common fission product.[66] The isotope's long half-life allows it to build up in spent nuclear fuel, reaching concentrations of up to several kilograms per ton of uranium in light-water reactors.[66] The decay sequence involves a combination of alpha and beta-minus decays, totaling seven alpha emissions and four beta emissions along the primary pathway to bismuth-209. Key intermediate nuclides include thorium-229 (half-life 7,340 years), which undergoes alpha decay, and shorter-lived species like protactinium-233 (half-life 26.97 days) and actinium-225 (half-life 9.92 days). The chain's progression reflects the typical actinide behavior, with alpha decay dominating mass number reduction while beta decay adjusts atomic numbers toward stability. Minor branching occurs at several points, including a 0.027% pathway at thorium-229 leading to radium-225 via an alternative route, though the dominant mode is direct alpha decay to radium-225. More notable branching is observed at francium-221 (beta-minus branch <0.1%) and especially at bismuth-213, where 2.09% of decays proceed via alpha emission to stable thallium-209 instead of the primary beta-minus path to polonium-213. These branches contribute negligibly to the overall chain flux but highlight the complexity of actinide decay networks. The following table summarizes the principal decay chain, including half-lives and dominant decay modes (branching ratios for minor paths are noted where significant):| Nuclide | Half-life | Decay mode | Daughter nuclide |
|---|---|---|---|
| ²³⁷Np | 2.144 × 10⁶ years | α | ²³³Pa |
| ²³³Pa | 26.97 days | β⁻ | ²³³U |
| ²³³U | 1.592 × 10⁵ years | α | ²²⁹Th |
| ²²⁹Th | 7,340 years | α | ²²⁵Ra |
| ²²⁵Ra | 14.9 days | β⁻ | ²²⁵Ac |
| ²²⁵Ac | 9.92 days | α | ²²¹Fr |
| ²²¹Fr | 4.8 minutes | α (β⁻ <0.1%) | ²¹⁷At |
| ²¹⁷At | 32.3 ms | α (β⁻ 0.01%) | ²¹³Bi |
| ²¹³Bi | 45.59 minutes | β⁻ (97.91%); α (2.09%) | ²¹³Po (main); ²⁰⁹Tl (branch) |
| ²¹³Po | 4.2 μs | α | ²⁰⁹Pb |
| ²⁰⁹Pb | 3.25 hours | β⁻ | ²⁰⁹Bi (stable) |