
Community hub
Recent from talks
Contribute something to knowledge base
Content stats: 0 posts, 0 articles, 1 media, 0 notes
Members stats: 0 subscribers, 0 contributors, 0 moderators, 0 supporters
Subscribers
Supporters
Contributors
Moderators
Hub AI
Protein isoform AI simulator
(@Protein isoform_simulator)
Hub AI
Protein isoform AI simulator
(@Protein isoform_simulator)
Protein isoform
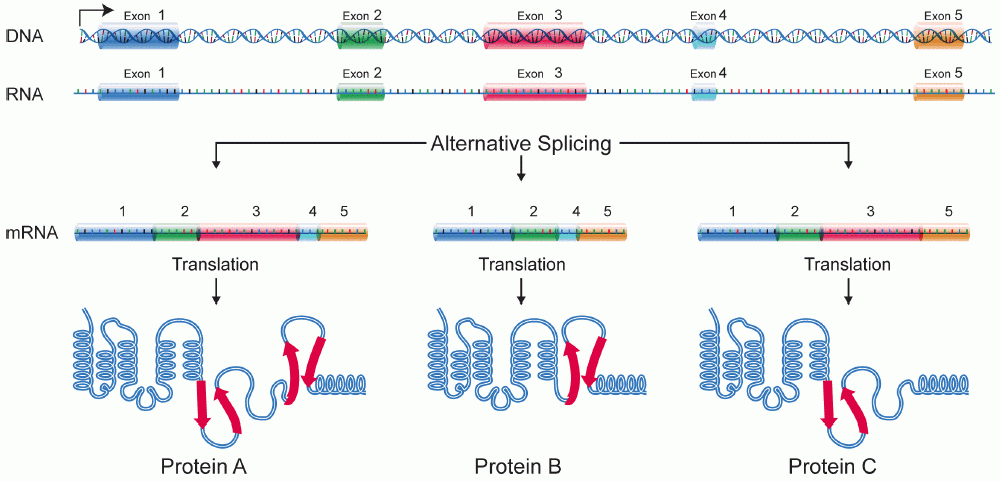
A protein isoform, or "protein variant", is a member of a set of highly similar proteins that originate from a single gene and are the result of genetic differences. While many perform the same or similar biological roles, some isoforms have unique functions. A set of protein isoforms may be formed from alternative splicings, variable promoter usage, or other post-transcriptional modifications of a single gene; post-translational modifications are generally not considered. (For that, see Proteoforms.) Through RNA splicing mechanisms, mRNA has the ability to select different protein-coding segments (exons) of a gene, or even different parts of exons from RNA to form different mRNA sequences. Each unique sequence produces a specific form of a protein.
The discovery of isoforms could explain the discrepancy between the small number of protein coding regions of genes revealed by the Human Genome Project and the large diversity of proteins seen in an organism: different proteins encoded by the same gene could increase the diversity of the proteome. Isoforms at the RNA level are readily characterized by cDNA transcript studies. Many human genes possess confirmed alternative splicing isoforms. It has been estimated that ~100,000 expressed sequence tags (ESTs) can be identified in humans. Isoforms at the protein level can manifest in the deletion of whole domains or shorter loops, usually located on the surface of the protein.
One single gene has the ability to produce multiple proteins that differ both in structure and composition; this process is regulated by the alternative splicing of mRNA, though it is not clear to what extent such a process affects the diversity of the human proteome, as the abundance of mRNA transcript isoforms does not necessarily correlate with the abundance of protein isoforms. Three-dimensional protein structure comparisons can be used to help determine which, if any, isoforms represent functional protein products, and the structure of most isoforms in the human proteome has been predicted by AlphaFold and publicly released at isoform.io. The specificity of translated isoforms is derived by the protein's structure/function, as well as the cell type and developmental stage during which they are produced. Determining specificity becomes more complicated when a protein has multiple subunits and each subunit has multiple isoforms.
For example, the 5' AMP-activated protein kinase (AMPK), an enzyme, which performs different roles in human cells, has 3 subunits:
In human skeletal muscle, the preferred form is α2β2γ1. But in the human liver, the most abundant form is α1β2γ1.
The primary mechanisms that produce protein isoforms are alternative splicing and variable promoter usage, though modifications due to genetic changes, such as mutations and polymorphisms are sometimes also considered distinct isoforms.
Alternative splicing is the main post-transcriptional modification process that produces mRNA transcript isoforms, and is a major molecular mechanism that may contribute to protein diversity. The spliceosome, a large ribonucleoprotein, is the molecular machine inside the nucleus responsible for RNA cleavage and ligation, removing non-protein coding segments (introns).
Because splicing is a process that occurs between transcription and translation, its primary effects have mainly been studied through genomics techniques—for example, microarray analyses and RNA sequencing have been used to identify alternatively spliced transcripts and measure their abundances. Transcript abundance is often used as a proxy for the abundance of protein isoforms, though proteomics experiments using gel electrophoresis and mass spectrometry have demonstrated that the correlation between transcript and protein counts is often low, and that one protein isoform is usually dominant. One 2015 study states that the cause of this discrepancy likely occurs after translation, though the mechanism is essentially unknown. Consequently, although alternative splicing has been implicated as an important link between variation and disease, there is no conclusive evidence that it acts primarily by producing novel protein isoforms.
Protein isoform
A protein isoform, or "protein variant", is a member of a set of highly similar proteins that originate from a single gene and are the result of genetic differences. While many perform the same or similar biological roles, some isoforms have unique functions. A set of protein isoforms may be formed from alternative splicings, variable promoter usage, or other post-transcriptional modifications of a single gene; post-translational modifications are generally not considered. (For that, see Proteoforms.) Through RNA splicing mechanisms, mRNA has the ability to select different protein-coding segments (exons) of a gene, or even different parts of exons from RNA to form different mRNA sequences. Each unique sequence produces a specific form of a protein.
The discovery of isoforms could explain the discrepancy between the small number of protein coding regions of genes revealed by the Human Genome Project and the large diversity of proteins seen in an organism: different proteins encoded by the same gene could increase the diversity of the proteome. Isoforms at the RNA level are readily characterized by cDNA transcript studies. Many human genes possess confirmed alternative splicing isoforms. It has been estimated that ~100,000 expressed sequence tags (ESTs) can be identified in humans. Isoforms at the protein level can manifest in the deletion of whole domains or shorter loops, usually located on the surface of the protein.
One single gene has the ability to produce multiple proteins that differ both in structure and composition; this process is regulated by the alternative splicing of mRNA, though it is not clear to what extent such a process affects the diversity of the human proteome, as the abundance of mRNA transcript isoforms does not necessarily correlate with the abundance of protein isoforms. Three-dimensional protein structure comparisons can be used to help determine which, if any, isoforms represent functional protein products, and the structure of most isoforms in the human proteome has been predicted by AlphaFold and publicly released at isoform.io. The specificity of translated isoforms is derived by the protein's structure/function, as well as the cell type and developmental stage during which they are produced. Determining specificity becomes more complicated when a protein has multiple subunits and each subunit has multiple isoforms.
For example, the 5' AMP-activated protein kinase (AMPK), an enzyme, which performs different roles in human cells, has 3 subunits:
In human skeletal muscle, the preferred form is α2β2γ1. But in the human liver, the most abundant form is α1β2γ1.
The primary mechanisms that produce protein isoforms are alternative splicing and variable promoter usage, though modifications due to genetic changes, such as mutations and polymorphisms are sometimes also considered distinct isoforms.
Alternative splicing is the main post-transcriptional modification process that produces mRNA transcript isoforms, and is a major molecular mechanism that may contribute to protein diversity. The spliceosome, a large ribonucleoprotein, is the molecular machine inside the nucleus responsible for RNA cleavage and ligation, removing non-protein coding segments (introns).
Because splicing is a process that occurs between transcription and translation, its primary effects have mainly been studied through genomics techniques—for example, microarray analyses and RNA sequencing have been used to identify alternatively spliced transcripts and measure their abundances. Transcript abundance is often used as a proxy for the abundance of protein isoforms, though proteomics experiments using gel electrophoresis and mass spectrometry have demonstrated that the correlation between transcript and protein counts is often low, and that one protein isoform is usually dominant. One 2015 study states that the cause of this discrepancy likely occurs after translation, though the mechanism is essentially unknown. Consequently, although alternative splicing has been implicated as an important link between variation and disease, there is no conclusive evidence that it acts primarily by producing novel protein isoforms.
Recent media
Recent media