Recent from talks
Fibronectin
Knowledge base stats:
Talk channels stats:
Members stats:
Fibronectin
Fibronectin is a high-molecular weight (~500-~600 kDa) glycoprotein of the extracellular matrix that binds to membrane-spanning receptor proteins called integrins. Fibronectin also binds to other extracellular matrix proteins such as collagen, fibrin, and heparan sulfate proteoglycans (e.g. syndecans).
Fibronectin exists as a protein dimer, consisting of two nearly identical monomers linked by a pair of disulfide bonds. The fibronectin protein is produced from a single gene, but alternative splicing of its pre-mRNA leads to the creation of several isoforms.
Two types of fibronectin are present in vertebrates:
Fibronectin plays a major role in cell adhesion, growth, migration, and differentiation, and it is important for processes such as wound healing and embryonic development. Altered fibronectin expression, degradation, and organization has been associated with a number of pathologies, including cancer, arthritis, and fibrosis.
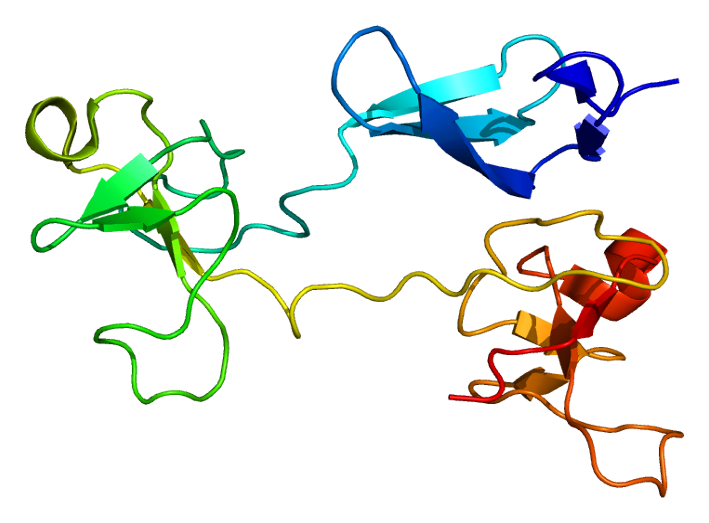
Fibronectin exists as a protein dimer, consisting of two nearly identical polypeptide chains linked by a pair of C-terminal disulfide bonds. Each fibronectin subunit has a molecular weight of ~230–~275 kDa and contains multiple copies of three types of modules: type I, II, and III. All three modules are composed of two anti-parallel β-sheets resulting in a Beta-sandwich; however, type I and type II are stabilized by intra-chain disulfide bonds, while type III modules do not contain any disulfide bonds. The absence of disulfide bonds in type III modules allows them to partially unfold under applied force.
Three regions of variable splicing occur along the length of the fibronectin protomer. One or both of the "extra" type III modules (EIIIA and EIIIB) may be present in cellular fibronectin, but they are never present in plasma fibronectin. A "variable" V-region exists between III14–15 (the 14th and 15th type III module). The V-region structure is different from the type I, II, and III modules, and its presence and length may vary. The V-region contains the binding site for α4β1 integrins. It is present in most cellular fibronectin, but only one of the two subunits in a plasma fibronectin dimer contains a V-region sequence.
The modules are arranged into several functional and protein-binding domains along the length of a fibronectin monomer. There are four fibronectin-binding domains, allowing fibronectin to associate with other fibronectin molecules. One of these fibronectin-binding domains, I1–5, is referred to as the "assembly domain", and it is required for the initiation of fibronectin matrix assembly. Modules III9–10 correspond to the "cell-binding domain" of fibronectin. The RGD sequence (Arg–Gly–Asp) is located in III10 and is the site of cell attachment via α5β1 and αVβ3 integrins on the cell surface. The "synergy site" is in III9 and has a role in modulating fibronectin's association with α5β1 integrins. Fibronectin also contains domains for fibrin-binding (I1–5, I10–12), collagen-binding (I6–9), fibulin-1-binding (III13–14), heparin-binding and syndecan-binding (III12–14).
Fibronectin has numerous functions that ensure the normal functioning of vertebrate organisms. It is involved in cell adhesion, growth, migration, and differentiation. Cellular fibronectin is assembled into the extracellular matrix, an insoluble network that separates and supports the organs and tissues of an organism.
Hub AI
Fibronectin AI simulator
(@Fibronectin_simulator)
Fibronectin
Fibronectin is a high-molecular weight (~500-~600 kDa) glycoprotein of the extracellular matrix that binds to membrane-spanning receptor proteins called integrins. Fibronectin also binds to other extracellular matrix proteins such as collagen, fibrin, and heparan sulfate proteoglycans (e.g. syndecans).
Fibronectin exists as a protein dimer, consisting of two nearly identical monomers linked by a pair of disulfide bonds. The fibronectin protein is produced from a single gene, but alternative splicing of its pre-mRNA leads to the creation of several isoforms.
Two types of fibronectin are present in vertebrates:
Fibronectin plays a major role in cell adhesion, growth, migration, and differentiation, and it is important for processes such as wound healing and embryonic development. Altered fibronectin expression, degradation, and organization has been associated with a number of pathologies, including cancer, arthritis, and fibrosis.
Fibronectin exists as a protein dimer, consisting of two nearly identical polypeptide chains linked by a pair of C-terminal disulfide bonds. Each fibronectin subunit has a molecular weight of ~230–~275 kDa and contains multiple copies of three types of modules: type I, II, and III. All three modules are composed of two anti-parallel β-sheets resulting in a Beta-sandwich; however, type I and type II are stabilized by intra-chain disulfide bonds, while type III modules do not contain any disulfide bonds. The absence of disulfide bonds in type III modules allows them to partially unfold under applied force.
Three regions of variable splicing occur along the length of the fibronectin protomer. One or both of the "extra" type III modules (EIIIA and EIIIB) may be present in cellular fibronectin, but they are never present in plasma fibronectin. A "variable" V-region exists between III14–15 (the 14th and 15th type III module). The V-region structure is different from the type I, II, and III modules, and its presence and length may vary. The V-region contains the binding site for α4β1 integrins. It is present in most cellular fibronectin, but only one of the two subunits in a plasma fibronectin dimer contains a V-region sequence.
The modules are arranged into several functional and protein-binding domains along the length of a fibronectin monomer. There are four fibronectin-binding domains, allowing fibronectin to associate with other fibronectin molecules. One of these fibronectin-binding domains, I1–5, is referred to as the "assembly domain", and it is required for the initiation of fibronectin matrix assembly. Modules III9–10 correspond to the "cell-binding domain" of fibronectin. The RGD sequence (Arg–Gly–Asp) is located in III10 and is the site of cell attachment via α5β1 and αVβ3 integrins on the cell surface. The "synergy site" is in III9 and has a role in modulating fibronectin's association with α5β1 integrins. Fibronectin also contains domains for fibrin-binding (I1–5, I10–12), collagen-binding (I6–9), fibulin-1-binding (III13–14), heparin-binding and syndecan-binding (III12–14).
Fibronectin has numerous functions that ensure the normal functioning of vertebrate organisms. It is involved in cell adhesion, growth, migration, and differentiation. Cellular fibronectin is assembled into the extracellular matrix, an insoluble network that separates and supports the organs and tissues of an organism.
Recent media