| Jmol | |
|---|---|
 Jmol three-dimensional structure rendering of streptavidin | |
| Developer | Jmol development team |
| Initial release | 2001 |
| Stable release | 16.3.7 |
| Repository | sourceforge |
| Written in | Java |
| Operating system | Cross-platform |
| Platform | Systems with Java and Web browsers without Java |
| Available in | 24 languages |
List of languages Basque, Catalan, Chinese (CN and TW), Czech, Danish, Dutch, English (GB and US), Finnish, French, German, Hungarian, Indonesian, Italian, Japanese, Korean, Malay, Portuguese (BR), Russian, Spanish, Swedish, Turkish and Ukrainian [1] | |
| Type | Molecular modelling |
| License | LGPL 2.0 |
| Website | www |
Jmol is computer software for molecular modelling of chemical structures in 3 dimensions.[2] It is an open-source Java viewer for chemical structures in 3D.[3] The name originated from [J]ava (the programming language) + [mol]ecules, and also the mol file format.
JSmol is an implementation in JavaScript of the functionality of Jmol.[4] It can hence be embedded in web pages to display interactive 3D models of molecules and other structures without the need for any software apart from the web browser (it does not use Java).
Both Jmol and JSmol render an interactive 3D representation of a molecule or other structure that may be used as a teaching tool,[5] or for research, in several fields, e.g. chemistry, biochemistry, materials science, crystallography,[6] symmetry or nanotechnology.
Software
[edit]Jmol is written in the programming language Java, so it can run on different operating systems: Windows, macOS, Linux, and Unix, as long as they have Java installed. It is free and open-source software released under the GNU Lesser General Public License (LGPL) version 2.0. The interface is translated into more than 20 languages.
There are several products implemented:
- A standalone application (the Jmol application), composed of a single
Jmol.jarfile that can be used without installation, requiring only that the computer has Java installed. - A software development kit (SDK), i.e. a component that can be integrated into other Java applications, such as Bioclipse and Taverna.
- JSmol, a JavaScript library that allows integration of the 3D models in web pages and wikis.
Molecules can be displayed in different styles of rendering, like ball-and-stick models, space-filling models, ribbon diagrams, molecular surfaces, etc.[7] Jmol supports a wide range of chemical file formats, including Protein Data Bank (pdb), Crystallographic Information File (cif and mmcif), MDL Molfile (mol and sdf), and Chemical Markup Language (CML). It can also display other types of files for structures with 3D data.
JSmol replaced the Jmol Java applet, which in turn had been previously developed as an alternative to the Chime plug-in,[5] both of which became unsupported by web browsers. Jmol was initiated[8] to reproduce functions present in Chime (with the exception of the Sculpt mode) and has been continuously growing in features, surpassing the simple display of molecular structures. Most notably, it has a large set of commands and a thorough scripting language (JmolScript)[9] that includes many characteristics of a programming language, such as variables, arrays, mathematical and Boolean operators, SQL-like queries, functions, loops, conditionals, try-catch, switch...
Screenshots
[edit]-
 Two translucent planes illustrating symmetry planes for the water molecule.
Two translucent planes illustrating symmetry planes for the water molecule. -
 The electrostatic potential of allene mapped onto a translucent surface.
The electrostatic potential of allene mapped onto a translucent surface. -
 A shaded rendering of caffeine, with some measurements shown (distance, angle, dihedral)
A shaded rendering of caffeine, with some measurements shown (distance, angle, dihedral) -
 An SN2 reaction animated displaying the distance measured between the chlorine and carbon atoms.
An SN2 reaction animated displaying the distance measured between the chlorine and carbon atoms. -
 Display of quartz crystal structure, filling 2×2×2 unit cells. The unit cell boundbox is drawn and the unit cell data are shown on upper left.
Display of quartz crystal structure, filling 2×2×2 unit cells. The unit cell boundbox is drawn and the unit cell data are shown on upper left. -
 Cartoon p-orbitals of benzene together with the corresponding translucent molecular orbital.
Cartoon p-orbitals of benzene together with the corresponding translucent molecular orbital. -
 Crystal structure of an H/ACA box RNP from Pyrococcus furiosus
Crystal structure of an H/ACA box RNP from Pyrococcus furiosus -
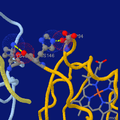 Highlighting two salt bridges in hemoglobin tetramer (hemo group as sticks at bottom-right)
Highlighting two salt bridges in hemoglobin tetramer (hemo group as sticks at bottom-right) -
 A fragment of transcription factor TFIIIA forming three consecutive zinc finger motifs, bound to a stretch of DNA
A fragment of transcription factor TFIIIA forming three consecutive zinc finger motifs, bound to a stretch of DNA -
 Eubacterial 70S Ribosome from Thermus thermophilus
Eubacterial 70S Ribosome from Thermus thermophilus










See also
[edit]References
[edit]- ^ Jmol translations
- ^ Chen, Jim X. (2008), Springer (ed.), Guide to Graphics Software Tools, p. 471, ISBN 978-1-84800-900-4
- ^ "How to cite Jmol".
- ^ "JSmol". Retrieved 2025-01-11.
- ^ a b Herráez, A (2006), "Biomolecules in the Computer: Jmol to the Rescue", Biochemistry and Molecular Biology Education, 34 (4): 255–61, doi:10.1002/bmb.2006.494034042644, PMID 21638687, S2CID 36319720
- ^ Hanson, Robert M. (2010), "Jmol – a paradigm shift in crystallographic visualization", Journal of Applied Crystallography, 43 (5): 1250–1260, doi:10.1107/S0021889810030256
- ^ Herráez, A (2007). How to Use Jmol to Study and Present Molecular Structures, Volume 1. Lulu Enterprises: Morrisville, NC, USA. p. 21. ISBN 978-1-84799-259-8.
- ^ Willighagen, Egon; Howard, Miguel (2007), "Fast and scriptable molecular graphics in web browsers without Java3D", Nature Precedings, doi:10.1038/npre.2007.50.1
- ^ "Jmol/JSmol Scripting Documentation".
External links
[edit]- Demonstration of JSmol capabilities. A simple page with interactive demonstrations of several features of JSmol embedded in the page.
- Jmol website (Official website, though not fully up to date)
- Jmol Wiki (active and updated) with, among other information, listings of websites, wikis, and moodles
- Hanson, Robert M.; Prilusky, Jaime; Renjian, Zhou; Nakane, Takanori; Sussman, Joel L. (2013). "JSmol and the next-generation web-based representation of 3D molecular structure as applied to Proteopedia". Israel Journal of Chemistry. doi:10.1002/ijch.201300024.
- Jmol extension at MediaWiki.
- Jmol extension for MediaWiki at SourceForge.
- Jmol extension for MediaWiki at JmolWiki.
- Proteopedia - Life in 3D, a collaborative and free 3D-encyclopedia of proteins and other biomolecules. (Uses Jmol Extension as a core component)
- Biomodel Complements for learning biochemistry and molecular biology.
- Molview
History
Origins and Early Development
Jmol originated in the late 1990s as an open-source initiative led by J. Daniel Gezelter to provide a free alternative to XMol, a proprietary molecular viewer developed at the Minnesota Supercomputer Center (now the Minnesota Supercomputer Institute). Gezelter, then a researcher focused on computational chemistry, initiated the project under the OpenScience umbrella to promote accessible scientific software without licensing restrictions or dependencies on closed-source tools. The primary motivation was to address the limitations of XMol, which had become obsolete and unavailable due to its proprietary nature, while enabling cross-platform 3D visualization of molecular structures using Java for broad compatibility across operating systems.[2][6] In 2002, Bradley A. Smith contributed significantly by streamlining the codebase, enhancing its usability and preparing it for wider adoption. Later that year, leadership transitioned to Egon Willighagen, who began efforts to integrate Jmol with the Chemical Development Kit (CDK), an open-source cheminformatics library, to improve handling of chemical structures and reactions; although full integration proved challenging due to performance concerns, this work laid groundwork for future interoperability. Concurrently, at the end of 2002, Michael T. Howard (known as Miguel) joined the project with the goal of positioning Jmol as a replacement for the Chime browser plugin, which was popular for web-based molecular viewing but tied to outdated technology.[2][7] Howard's involvement accelerated development, culminating in a major rewrite of the core classes in 2003 to boost performance and efficiency, particularly for rendering complex models without proprietary dependencies. This refocus emphasized Jmol's role as a lightweight, standalone Java application for basic 3D structure viewing, supporting educational and research needs in chemistry and biochemistry during its formative years. By the mid-2000s, these foundational efforts had established Jmol as a robust open-source tool, distinct from commercial alternatives.[2][8]Major Releases and Milestones
Jmol 10.0 was released in December 2004 as the first stable open-source replacement for the Chime plugin, featuring a high-performance graphics engine capable of handling large molecular structures.[2] This was followed by Jmol 10.2 in April 2006, which introduced refinements to the rendering system and improved file handling capabilities, enhancing overall usability through community testing.[2] In 2006, shortly after the 10.2 release, Bob Hanson assumed leadership of the Jmol development team, driving subsequent advancements.[2] Under Hanson's guidance, Jmol 11.0 launched in February 2007, marking a significant milestone with the addition of multi-file loading, advanced scripting functionalities, and specialized tools for crystallography, including support for surfaces and biopolymers.[2] Post-2007 development emphasized community-driven updates, with the project operating under the GNU Lesser General Public License (LGPL) to facilitate broader contributions and integration.[2][9] The software continued to evolve through incremental releases, culminating in version 16.3.35 in November 2025, which incorporated enhancements in performance optimization and cross-platform compatibility.[10]Features
Visualization and Rendering Capabilities
Jmol employs a custom Java-based graphics engine utilizing a z-buffer algorithm for 3D rendering, enabling efficient depth sorting and occlusion handling without reliance on hardware-accelerated APIs like OpenGL or Java3D. This pipeline constructs images offscreen before a single transfer to the display, optimizing performance on standard hardware while supporting high-resolution outputs. The engine facilitates interactive manipulation of molecular structures, including real-time rotations and zooms, and is extensible through scripting for dynamic visualizations.[11] Central to Jmol's visualization are diverse display models for representing atoms, bonds, and molecular architectures. Atoms and bonds can be rendered in ball-and-stick format by combining partial spacefilling spheres for atoms with cylindrical wireframe bonds, providing clear depiction of connectivity and van der Waals interactions. Space-filling models use full atomic radii to illustrate molecular volumes, while for biomolecules like proteins, ribbon diagrams—including cartoon, ribbon, and rocket styles—highlight secondary structures such as alpha helices and beta sheets. These models extend to crystals via unit cell overlays and to materials through periodic boundary visualizations, allowing users to toggle between representations for conceptual clarity.[12][13][14] Advanced rendering options enhance visual fidelity and interpretability. Perspective depth simulates realistic 3D projection, while antialiasing smooths edges to reduce jagged artifacts in both on-screen displays and exported images. Lighting effects include adjustable ambient, diffuse, and specular components, enabling shadowed and highlighted surfaces for better depth perception in complex structures. Transparency controls allow semi-opaque rendering of surfaces or chains, facilitating overlap analysis in biomolecules or crystal lattices. Isosurfaces and molecular orbitals can be generated and colored by properties like electron density or partial charges, supporting detailed examination of electronic structures.[15][16][17] For large structures exceeding thousands of atoms, Jmol incorporates performance optimizations within Java's graphics framework, such as level-of-detail adjustments via reduced mesh resolution for distant surfaces and selective hiding of non-essential elements like hydrogens. These techniques maintain interactive frame rates by minimizing polygon counts and leveraging efficient bounding volume hierarchies for culling. The engine's software-based approach ensures cross-platform consistency, though it benefits from system RAM for caching large datasets.[11][15] Jmol supports fluid animations through frame sequencing and keyframe interpolation, enabling vibrations, conformational changes, or trajectory playback. Rotations occur via quaternion-based transformations for smooth, axis-aligned or arbitrary motions. In-viewer measurements compute and label bond lengths, angles, torsions, and distances in real-time, with options for hydrogen bonds or van der Waals contacts, aiding quantitative structural analysis. These capabilities integrate with scripting for automated, dynamic presentations.[18][19][20]Supported File Formats and Data Handling
Jmol supports a wide array of input file formats for importing chemical structures and data, enabling users to load molecular models from various sources without extensive preprocessing. Key formats include the Protein Data Bank (PDB) format for biomolecular structures, Crystallographic Information File (CIF) and macromolecular CIF (mmCIF) for crystallographic data, MDL Molfile (MOL) and SDF for single or multiple molecular records, Gaussian output files for quantum chemistry calculations, and GAMESS log files for similar computational results.[1][21] The software automatically decompresses gzip-compressed files upon loading, facilitating the handling of archived datasets commonly distributed in scientific repositories. Additionally, Jmol accommodates multi-model datasets, such as molecular dynamics trajectories or structural ensembles in SDF or PDB formats, allowing sequential access to frames via scripting or navigation commands for animation and analysis.[1] For output, Jmol provides export capabilities to several formats that preserve visual or structural information. Images can be saved as PNG files, which may include embedded molecular state data in PNGJ format for later restoration. High-quality ray-traced renderings are supported through export to POV-Ray scene description files, enabling further processing in dedicated rendering software. Scripted state files capture the current session configuration, including loaded models and viewing parameters, for reproducibility. Structural data can also be exported to MOL format for interoperability with other chemistry tools.[22][23] In terms of data integrity, Jmol includes mechanisms for error handling during file parsing, such as an option to ignore unrecognized sections in files while attempting to extract valid model data, which aids in processing partially malformed or mixed-format inputs. For format conversions, Jmol integrates utilities to transform loaded data between supported types, such as generating 3D coordinates from SMILES strings via external web services or exporting crystal structures to printable formats like STL, though primary conversions rely on internal parsing rather than full format transmutation.[21][24]Scripting and Automation
Jmol's scripting language provides a powerful mechanism for automating molecular visualizations and analyses, building on a RasMol-like command syntax while incorporating JavaScript-style programming elements for enhanced flexibility.[25] This allows users to create complex scripts for tasks ranging from simple model manipulations to intricate computational workflows, with major enhancements introduced in version 11.0, including user-defined functions, variables, and control structures.[2] The core syntax resembles RasMol commands, such asload for file import and select for atom selection, but extends to support variables, loops, conditionals, and JmolMath functions for advanced calculations. Variables can be global or local, supporting types like integers, decimals, strings, arrays, bitsets, points, and planes; for example, var x = 10 declares an integer variable.[25] Control structures include if/else for conditionals (e.g., if (x > 0) { print x } else { print "negative" }), for loops (e.g., for (var i = 1; i < 10; i++) { ... }), and while loops, enabling iterative processing of molecular data.[25]
JmolMath functions facilitate geometric and analytical operations, such as vector cross products via cross(toAtom, toUser) or distance measurements with x.distance({0 0 0}) for a point's distance to the origin.[25] Model manipulation commands include select (e.g., select carbon to isolate carbon atoms), color (e.g., color atoms green), and rotate (e.g., rotate y 90 for a 90-degree rotation around the y-axis). Animation scripting uses commands like animation on and frame 1 to control frame sequences, while integration with external tools occurs through callbacks, such as set callback echo for script status feedback.[25]
State scripting enables saving and restoring sessions with save state myState and restore state myState, preserving model views, selections, and parameters for reproducible workflows. Menu and button automation allows interactive environments via menu (e.g., menu "Rotate" rotate y 90) to create custom controls that execute scripts on user input.[25]
Practical examples illustrate scripting's utility: for hydrogen addition, the command calculate hydrogens infers and adds hydrogens to a loaded model; for surface generation, isosurface solvent accessible computes a solvent-accessible surface, which can be colored or animated within a larger script.[25] These features make Jmol scripting ideal for automating repetitive tasks in education and research, such as batch processing of molecular structures or dynamic visualizations.[25]
Implementations
Standalone Application
Jmol is distributed as a standalone Java application that operates independently on desktop systems, enabling users to visualize and manipulate 3D molecular structures without requiring a web browser. The application is cross-platform compatible, running on Windows, macOS, and Linux operating systems as long as a Java Runtime Environment (JRE) version 1.4 or higher is installed on the host machine.[14][26] To deploy it, users download a single JAR file from the official repository, which can be executed directly by double-clicking or via command line with a Java interpreter, eliminating the need for complex installation procedures.[26] The user interface of the standalone Jmol application includes a menu bar at the top for essential file operations, such as opening molecular files in supported formats, saving sessions, and accessing the script console. The console provides an interactive input area for entering Jmol scripting commands to customize visualizations, while the toolbar offers quick-access buttons for frequent tasks like zooming, rotating the model, and selecting rendering styles such as ball-and-stick or space-filling representations.[27][28][29] As a desktop program, Jmol excels in offline scenarios, permitting the loading of local structure files without network access, the execution of pre-written scripts for automated animations or analyses, and the export of outputs including high-resolution static images in formats like PNG or animated movies in AVI or GIF.[1][26] System requirements remain minimal, with no specialized hardware beyond a standard graphics card, though the application recommends using the latest available Java version for optimal performance and security; Java 1.4 is the stated minimum, but versions prior to that are unsupported.[14] For graphics-related issues, such as jagged edges from lack of default anti-aliasing or slowdowns in large viewing windows due to increased pixel rendering, users can troubleshoot by enabling anti-aliasing through console commands likeset antialiasDisplay true or adjusting Java's hardware acceleration settings in the control panel.[14][30]