Recent from talks
Osteosclerosis
Knowledge base stats:
Talk channels stats:
Members stats:
Osteosclerosis
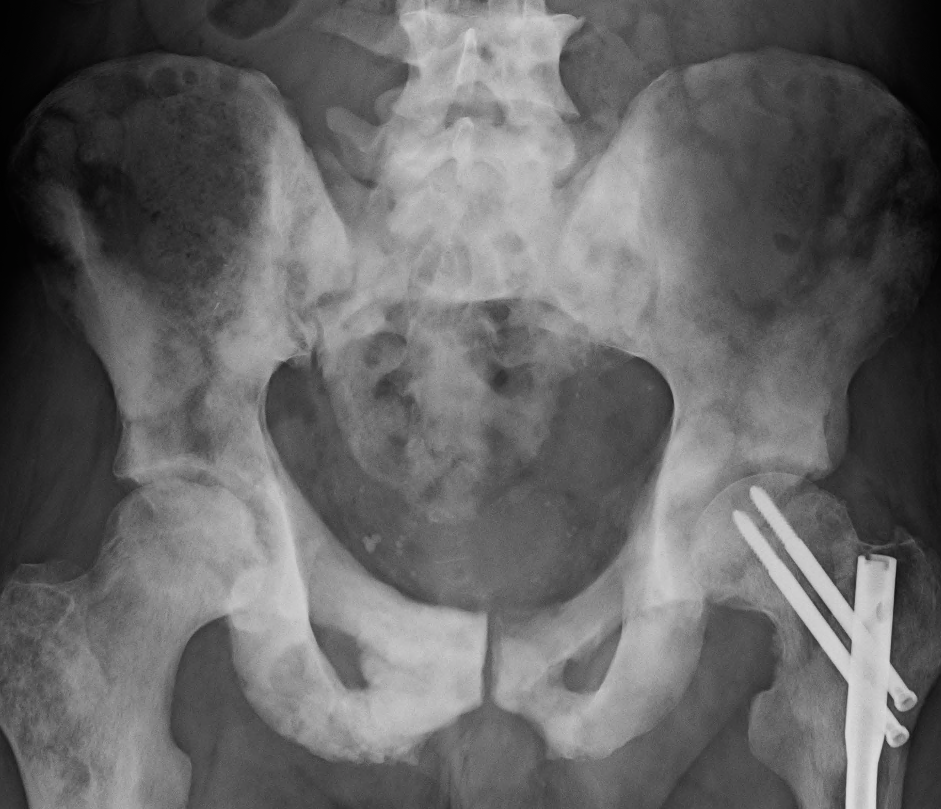
Osteosclerosis is a disorder characterized by abnormal hardening of bone and an elevation in bone density. It may predominantly affect the medullary portion and/or cortex of bone. Plain radiographs are a valuable tool for detecting and classifying osteosclerotic disorders. It can manifest in localized or generalized osteosclerosis. Localized osteosclerosis can be caused by Legg–Calvé–Perthes disease, sickle-cell disease and osteoarthritis among others. Osteosclerosis can be classified in accordance with the causative factor into acquired and hereditary.
Osteosclerosis can be detected with a simple radiography. There are white portions of the bone which appear due to the increased number of bone trabeculae.[citation needed]
In the animal kingdom, there also exists a non-pathological form of osteosclerosis, resulting in unusually solid bone structure with little to no marrow. It is often seen in aquatic vertebrates, especially those living in shallow waters, providing ballast as an adaptation for an aquatic lifestyle. It makes bones heavier, but also more fragile. In those animal groups, osteosclerosis often occurs together with bone thickening (pachyostosis). This joint occurrence is called pachyosteosclerosis.
Hub AI
Osteosclerosis AI simulator
(@Osteosclerosis_simulator)
Osteosclerosis
Osteosclerosis is a disorder characterized by abnormal hardening of bone and an elevation in bone density. It may predominantly affect the medullary portion and/or cortex of bone. Plain radiographs are a valuable tool for detecting and classifying osteosclerotic disorders. It can manifest in localized or generalized osteosclerosis. Localized osteosclerosis can be caused by Legg–Calvé–Perthes disease, sickle-cell disease and osteoarthritis among others. Osteosclerosis can be classified in accordance with the causative factor into acquired and hereditary.
Osteosclerosis can be detected with a simple radiography. There are white portions of the bone which appear due to the increased number of bone trabeculae.[citation needed]
In the animal kingdom, there also exists a non-pathological form of osteosclerosis, resulting in unusually solid bone structure with little to no marrow. It is often seen in aquatic vertebrates, especially those living in shallow waters, providing ballast as an adaptation for an aquatic lifestyle. It makes bones heavier, but also more fragile. In those animal groups, osteosclerosis often occurs together with bone thickening (pachyostosis). This joint occurrence is called pachyosteosclerosis.
Recent media