Recent from talks
Pseudocapacitance
Knowledge base stats:
Talk channels stats:
Members stats:
Pseudocapacitance
Pseudocapacitance is the electrochemical storage of electricity in an electrochemical capacitor that occurs due to faradaic charge transfer originating from a very fast sequence of reversible faradaic redox, electrosorption or intercalation processes on the surface of suitable electrodes. Pseudocapacitance is accompanied by an electron charge-transfer between electrolyte and electrode coming from a de-solvated and adsorbed ion. One electron per charge unit is involved. The adsorbed ion has no chemical reaction with the atoms of the electrode (no chemical bonds arise) since only a charge-transfer takes place. Supercapacitors that rely primarily on pseudocapacitance are sometimes called pseudocapacitors.
Faradaic pseudocapacitance only occurs together with static double-layer capacitance. Pseudocapacitance and double-layer capacitance both contribute inseparably to the total capacitance value. The amount of pseudocapacitance depends on the surface area, material and structure of the electrodes. Pseudocapacitance may contribute more capacitance than double-layer capacitance for the same surface area by 100x.
The amount of electric charge stored in a pseudocapacitance is linearly proportional to the applied voltage. The unit of pseudocapacitance is farad.
Redox reactions in batteries with faradaic charge-transfer between an electrolyte and the surface of an electrode were characterized decades ago. These chemical processes are associated with chemical reactions of the electrode materials usually with attendant phase changes. Although these chemical processes are relatively reversible, battery charge/discharge cycles often irreversibly produce unreversed chemical reaction products of the reagents. Accordingly, the cycle-life of rechargeable batteries is usually limited. Further, the reaction products lower power density. Additionally, the chemical processes are relatively slow, extending charge/discharge times.
A fundamental difference between redox reactions in batteries and in electrochemical capacitors (supercapacitors) is that in the latter, the reactions are a very fast sequence of reversible processes with electron transfer without any phase changes of the electrode molecules. They do not involve making or breaking chemical bonds. The de-solvated atoms or ions contributing the pseudocapacitance simply cling to the atomic structure of the electrode and charges are distributed on surfaces by physical adsorption processes. Compared with batteries, supercapacitor faradaic processes are much faster and more stable over time, because they leave only traces of reaction products. Despite the reduced amount of these products, they cause capacitance degradation. This behavior is the essence of pseudocapacitance.
Pseudocapacitive processes lead to a charge-dependent, linear capacitive behavior, as well as the accomplishment of non-faradaic double-layer capacitance in contrast to batteries, which have a nearly charge-independent behavior. The amount of pseudocapacitance depends on the surface area, material and structure of the electrodes. The pseudocapacitance may exceed the value of double-layer capacitance for the same surface area by 100x.
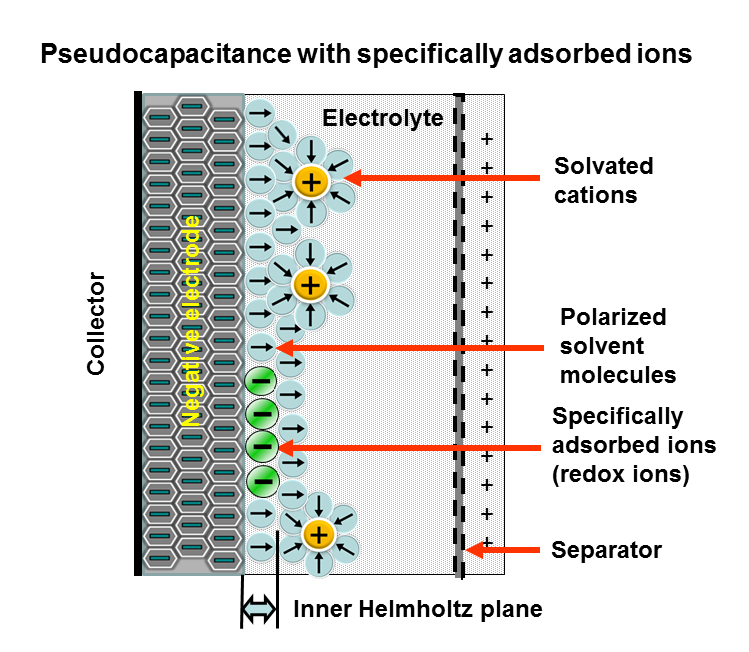
Applying a voltage at the capacitor terminals moves the polarized ions or charged atoms in the electrolyte to the opposite polarized electrode. Between the surfaces of the electrodes and the adjacent electrolyte an electric double-layer forms. One layer of ions on the electrode surface and the second layer of adjacent polarized and solvated ions in the electrolyte move to the opposite polarized electrode. The two ion layers are separated by a single layer of electrolyte molecules. Between the two layers, a static electric field forms that results in double-layer capacitance. Accompanied by the electric double-layer, some de-solvated electrolyte ions pervade the separating solvent layer and are adsorbed by the electrode's surface atoms. They are specifically adsorbed and deliver their charge to the electrode. In other words, the ions in the electrolyte within the Helmholtz double-layer also act as electron donors and transfer electrons to the electrode atoms, resulting in a faradaic current. This faradaic charge transfer, originated by a fast sequence of reversible redox reactions, electrosorptions or intercalation processes between electrolyte and the electrode surface is called pseudocapacitance.
Depending on the electrode's structure or surface material, pseudocapacitance can originate when specifically adsorbed ions pervade the double-layer, proceeding in several one-electron stages. The electrons involved in the faradaic processes are transferred to or from the electrode's valence-electron states (orbitals) and flow through the external circuit to the opposite electrode where a second double-layer with an equal number of opposite-charged ions forms. The electrons remain in the strongly ionized and electrode surface's "electron hungry" transition-metal ions and are not transferred to the adsorbed ions. This kind of pseudocapacitance has a linear function within narrow limits and is determined by the potential-dependent degree of surface coverage of the adsorbed anions. The storage capacity of the pseudocapacitance is limited by the finite quantity of reagent or of available surface.
Hub AI
Pseudocapacitance AI simulator
(@Pseudocapacitance_simulator)
Pseudocapacitance
Pseudocapacitance is the electrochemical storage of electricity in an electrochemical capacitor that occurs due to faradaic charge transfer originating from a very fast sequence of reversible faradaic redox, electrosorption or intercalation processes on the surface of suitable electrodes. Pseudocapacitance is accompanied by an electron charge-transfer between electrolyte and electrode coming from a de-solvated and adsorbed ion. One electron per charge unit is involved. The adsorbed ion has no chemical reaction with the atoms of the electrode (no chemical bonds arise) since only a charge-transfer takes place. Supercapacitors that rely primarily on pseudocapacitance are sometimes called pseudocapacitors.
Faradaic pseudocapacitance only occurs together with static double-layer capacitance. Pseudocapacitance and double-layer capacitance both contribute inseparably to the total capacitance value. The amount of pseudocapacitance depends on the surface area, material and structure of the electrodes. Pseudocapacitance may contribute more capacitance than double-layer capacitance for the same surface area by 100x.
The amount of electric charge stored in a pseudocapacitance is linearly proportional to the applied voltage. The unit of pseudocapacitance is farad.
Redox reactions in batteries with faradaic charge-transfer between an electrolyte and the surface of an electrode were characterized decades ago. These chemical processes are associated with chemical reactions of the electrode materials usually with attendant phase changes. Although these chemical processes are relatively reversible, battery charge/discharge cycles often irreversibly produce unreversed chemical reaction products of the reagents. Accordingly, the cycle-life of rechargeable batteries is usually limited. Further, the reaction products lower power density. Additionally, the chemical processes are relatively slow, extending charge/discharge times.
A fundamental difference between redox reactions in batteries and in electrochemical capacitors (supercapacitors) is that in the latter, the reactions are a very fast sequence of reversible processes with electron transfer without any phase changes of the electrode molecules. They do not involve making or breaking chemical bonds. The de-solvated atoms or ions contributing the pseudocapacitance simply cling to the atomic structure of the electrode and charges are distributed on surfaces by physical adsorption processes. Compared with batteries, supercapacitor faradaic processes are much faster and more stable over time, because they leave only traces of reaction products. Despite the reduced amount of these products, they cause capacitance degradation. This behavior is the essence of pseudocapacitance.
Pseudocapacitive processes lead to a charge-dependent, linear capacitive behavior, as well as the accomplishment of non-faradaic double-layer capacitance in contrast to batteries, which have a nearly charge-independent behavior. The amount of pseudocapacitance depends on the surface area, material and structure of the electrodes. The pseudocapacitance may exceed the value of double-layer capacitance for the same surface area by 100x.
Applying a voltage at the capacitor terminals moves the polarized ions or charged atoms in the electrolyte to the opposite polarized electrode. Between the surfaces of the electrodes and the adjacent electrolyte an electric double-layer forms. One layer of ions on the electrode surface and the second layer of adjacent polarized and solvated ions in the electrolyte move to the opposite polarized electrode. The two ion layers are separated by a single layer of electrolyte molecules. Between the two layers, a static electric field forms that results in double-layer capacitance. Accompanied by the electric double-layer, some de-solvated electrolyte ions pervade the separating solvent layer and are adsorbed by the electrode's surface atoms. They are specifically adsorbed and deliver their charge to the electrode. In other words, the ions in the electrolyte within the Helmholtz double-layer also act as electron donors and transfer electrons to the electrode atoms, resulting in a faradaic current. This faradaic charge transfer, originated by a fast sequence of reversible redox reactions, electrosorptions or intercalation processes between electrolyte and the electrode surface is called pseudocapacitance.
Depending on the electrode's structure or surface material, pseudocapacitance can originate when specifically adsorbed ions pervade the double-layer, proceeding in several one-electron stages. The electrons involved in the faradaic processes are transferred to or from the electrode's valence-electron states (orbitals) and flow through the external circuit to the opposite electrode where a second double-layer with an equal number of opposite-charged ions forms. The electrons remain in the strongly ionized and electrode surface's "electron hungry" transition-metal ions and are not transferred to the adsorbed ions. This kind of pseudocapacitance has a linear function within narrow limits and is determined by the potential-dependent degree of surface coverage of the adsorbed anions. The storage capacity of the pseudocapacitance is limited by the finite quantity of reagent or of available surface.
Recent media