
Community hub
Recent from talks
Contribute something
Nothing was collected or created yet.
Beta sheet
View on Wikipedia

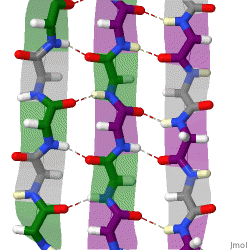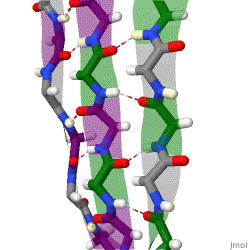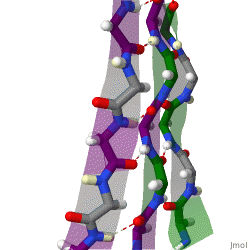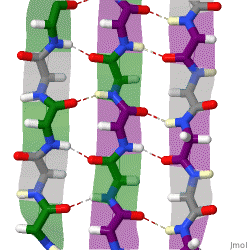
The beta sheet (β-sheet, also β-pleated sheet) is a common motif of the regular protein secondary structure. Beta sheets consist of beta strands (β-strands) connected laterally by at least two or three backbone hydrogen bonds, forming a generally twisted, pleated sheet. A β-strand is a stretch of polypeptide chain typically 3 to 10 amino acids long with backbone in an extended conformation. The supramolecular association of β-sheets has been implicated in the formation of the fibrils and protein aggregates observed in amyloidosis, Alzheimer's disease and other proteinopathies.
History
[edit]
The first β-sheet structure was proposed by William Astbury in the 1930s. He proposed the idea of hydrogen bonding between the peptide bonds of parallel or antiparallel extended β-strands. However, Astbury did not have the necessary data on the bond geometry of the amino acids in order to build accurate models, especially since he did not then know that the peptide bond was planar. A refined version was proposed by Linus Pauling and Robert Corey in 1951. Their model incorporated the planarity of the peptide bond which they previously explained as resulting from keto-enol tautomerization.
Structure and orientation
[edit]Geometry
[edit]The majority of β-strands are arranged adjacent to other strands and form an extensive hydrogen bond network with their neighbors in which the N−H groups in the backbone of one strand establish hydrogen bonds with the C=O groups in the backbone of the adjacent strands. In the fully extended β-strand, successive side chains point straight up and straight down in an alternating pattern. Adjacent β-strands in a β-sheet are aligned so that their Cα atoms are adjacent and their side chains point in the same direction. The "pleated" appearance of β-strands arises from tetrahedral chemical bonding at the Cα atom; for example, if a side chain points straight up, then the bonds to the C′ must point slightly downwards, since its bond angle is approximately 109.5°. The pleating causes the distance between Cα
i and Cα
i + 2 to be approximately 6 Å (0.60 nm), rather than the 7.6 Å (0.76 nm) expected from two fully extended trans peptides. The "sideways" distance between adjacent Cα atoms in hydrogen-bonded β-strands is roughly 5 Å (0.50 nm).

However, β-strands are rarely perfectly extended; rather, they exhibit a twist. The energetically preferred dihedral angles near (φ, ψ) = (–135°, 135°) (broadly, the upper left region of the Ramachandran plot) diverge significantly from the fully extended conformation (φ, ψ) = (–180°, 180°).[2] The twist is often associated with alternating fluctuations in the dihedral angles to prevent the individual β-strands in a larger sheet from splaying apart. A good example of a strongly twisted β-hairpin can be seen in the protein BPTI.
The side chains point outwards from the folds of the pleats, roughly perpendicularly to the plane of the sheet; successive amino acid residues point outwards on alternating faces of the sheet.
Hydrogen bonding patterns
[edit]

Because peptide chains have a directionality conferred by their N-terminus and C-terminus, β-strands too can be said to be directional. They are usually represented in protein topology diagrams by an arrow pointing toward the C-terminus. Adjacent β-strands can form hydrogen bonds in antiparallel, parallel, or mixed arrangements.
In an antiparallel arrangement, the successive β-strands alternate directions so that the N-terminus of one strand is adjacent to the C-terminus of the next. This is the arrangement that produces the strongest inter-strand stability because it allows the inter-strand hydrogen bonds between carbonyls and amines to be planar, which is their preferred orientation. The peptide backbone dihedral angles (φ, ψ) are about (–140°, 135°) in antiparallel sheets. In this case, if two atoms Cα
i and Cα
j are adjacent in two hydrogen-bonded β-strands, then they form two mutual backbone hydrogen bonds to each other's flanking peptide groups; this is known as a close pair of hydrogen bonds.
In a parallel arrangement, all of the N-termini of successive strands are oriented in the same direction; this orientation may be slightly less stable because it introduces nonplanarity in the inter-strand hydrogen bonding pattern. The dihedral angles (φ, ψ) are about (–120°, 115°) in parallel sheets. It is rare to find less than five interacting parallel strands in a motif, suggesting that a smaller number of strands may be unstable, however it is also fundamentally more difficult for parallel β-sheets to form because strands with N and C termini aligned necessarily must be very distant in sequence [citation needed]. There is also evidence that parallel β-sheet may be more stable since small amyloidogenic sequences appear to generally aggregate into β-sheet fibrils composed of primarily parallel β-sheet strands, where one would expect anti-parallel fibrils if anti-parallel were more stable.
In parallel β-sheet structure, if two atoms Cα
i and Cα
j are adjacent in two hydrogen-bonded β-strands, then they do not hydrogen bond to each other; rather, one residue forms hydrogen bonds to the residues that flank the other (but not vice versa). For example, residue i may form hydrogen bonds to residues j − 1 and j + 1; this is known as a wide pair of hydrogen bonds. By contrast, residue j may hydrogen-bond to different residues altogether, or to none at all.
The hydrogen bond arrangement in parallel beta sheet resembles that in an amide ring motif with 11 atoms.
Finally, an individual strand may exhibit a mixed bonding pattern, with a parallel strand on one side and an antiparallel strand on the other. Such arrangements are less common than a random distribution of orientations would suggest, suggesting that this pattern is less stable than the anti-parallel arrangement, however bioinformatic analysis always struggles with extracting structural thermodynamics since there are always numerous other structural features present in whole proteins. Also proteins are inherently constrained by folding kinetics as well as folding thermodynamics, so one must always be careful in concluding stability from bioinformatic analysis.
The hydrogen bonding of β-strands need not be perfect, but can exhibit localized disruptions known as β-bulges.
The hydrogen bonds lie roughly in the plane of the sheet, with the peptide carbonyl groups pointing in alternating directions with successive residues; for comparison, successive carbonyls point in the same direction in the alpha helix.
Amino acid propensities
[edit]Large aromatic residues (tyrosine, phenylalanine, tryptophan) and β-branched amino acids (threonine, valine, isoleucine) are favored to be found in β-strands in the middle of β-sheets. Different types of residues (such as proline) are likely to be found in the edge strands in β-sheets, presumably to avoid the "edge-to-edge" association between proteins that might lead to aggregation and amyloid formation.[3]
Common structural motifs
[edit]

β-hairpin motif
[edit]A very simple structural motif involving β-strands is the β-hairpin, in which two antiparallel strands are linked by a short loop of two to five residues, of which one is frequently a glycine or a proline, both of which can assume the dihedral-angle conformations required for a tight turn or a β-bulge loop. Individual strands can also be linked in more elaborate ways with longer loops that may contain α-helices.
Greek key motif
[edit]The Greek key motif consists of four adjacent antiparallel strands and their linking loops. It consists of three antiparallel strands connected by hairpins, while the fourth is adjacent to the first and linked to the third by a longer loop. This type of structure forms easily during the protein folding process.[4][5] It was named after a pattern common to Greek ornamental artwork (see meander).
β-α-β motif
[edit]Due to the chirality of their component amino acids, all strands exhibit right-handed twist evident in most higher-order β-sheet structures. In particular, the linking loop between two parallel strands almost always has a right-handed crossover chirality, which is strongly favored by the inherent twist of the sheet.[6] This linking loop frequently contains a helical region, in which case it is called a β-α-β motif. A closely related motif called a β-α-β-α motif forms the basic component of the most commonly observed protein tertiary structure, the TIM barrel.


β-meander motif
[edit]A simple supersecondary protein topology composed of two or more consecutive antiparallel β-strands linked together by hairpin loops.[8][9] This motif is common in β-sheets and can be found in several structural architectures including β-barrels and β-propellers.
The vast majority of β-meander regions in proteins are found packed against other motifs or sections of the polypeptide chain, forming portions of the hydrophobic core that canonically drives formation of the folded structure.[10] However, several notable exceptions include the Outer Surface Protein A (OspA) variants[7] and the Single Layer β-sheet Proteins (SLBPs)[11] which contain single-layer β-sheets in the absence of a traditional hydrophobic core. These β-rich proteins feature an extended single-layer β-meander β-sheets that are primarily stabilized via inter-β-strand interactions and hydrophobic interactions present in the turn regions connecting individual strands.
Psi-loop motif
[edit]The psi-loop (Ψ-loop) motif consists of two antiparallel strands with one strand in between that is connected to both by hydrogen bonds.[12] There are four possible strand topologies for single Ψ-loops.[13] This motif is rare as the process resulting in its formation seems unlikely to occur during protein folding. The Ψ-loop was first identified in the aspartic protease family.[13]
Structural architectures of proteins with β-sheets
[edit]β-sheets are present in all-β, α+β and α/β domains, and in many peptides or small proteins with poorly defined overall architecture.[14][15] All-β domains may form β-barrels, β-sandwiches, β-prisms, β-propellers, and β-helices.
Structural topology
[edit]The topology of a β-sheet describes the order of hydrogen-bonded β-strands along the backbone. For example, the flavodoxin fold has a five-stranded, parallel β-sheet with topology 21345; thus, the edge strands are β-strand 2 and β-strand 5 along the backbone. Spelled out explicitly, β-strand 2 is H-bonded to β-strand 1, which is H-bonded to β-strand 3, which is H-bonded to β-strand 4, which is H-bonded to β-strand 5, the other edge strand. In the same system, the Greek key motif described above has a 4123 topology. The secondary structure of a β-sheet can be described roughly by giving the number of strands, their topology, and whether their hydrogen bonds are parallel or antiparallel.
β-sheets can be open, meaning that they have two edge strands (as in the flavodoxin fold or the immunoglobulin fold) or they can be closed β-barrels (such as the TIM barrel). β-Barrels are often described by their stagger or shear. Some open β-sheets are very curved and fold over on themselves (as in the SH3 domain) or form horseshoe shapes (as in the ribonuclease inhibitor). Open β-sheets can assemble face-to-face (such as the β-propeller domain or immunoglobulin fold) or edge-to-edge, forming one big β-sheet.
Dynamic features
[edit]β-pleated sheet structures are made from extended β-strand polypeptide chains, with strands linked to their neighbours by hydrogen bonds. Due to this extended backbone conformation, β-sheets resist stretching. β-sheets in proteins may carry out low-frequency accordion-like motion as observed by the Raman spectroscopy[16] and analyzed with the quasi-continuum model.[17]
Parallel β-helices
[edit]
A β-helix is formed from repeating structural units consisting of two or three short β-strands linked by short loops. These units "stack" atop one another in a helical fashion so that successive repetitions of the same strand hydrogen-bond with each other in a parallel orientation. See the β-helix article for further information.
In lefthanded β-helices, the strands themselves are quite straight and untwisted; the resulting helical surfaces are nearly flat, forming a regular triangular prism shape, as shown for the 1QRE archaeal carbonic anhydrase at right. Other examples are the lipid A synthesis enzyme LpxA and insect antifreeze proteins with a regular array of Thr sidechains on one face that mimic the structure of ice.[18]

Righthanded β-helices, typified by the pectate lyase enzyme shown at left or P22 phage tailspike protein, have a less regular cross-section, longer and indented on one of the sides; of the three linker loops, one is consistently just two residues long and the others are variable, often elaborated to form a binding or active site.[19]
A two-sided β-helix (right-handed) is found in some bacterial metalloproteases; its two loops are each six residues long and bind stabilizing calcium ions to maintain the integrity of the structure, using the backbone and the Asp side chain oxygens of a GGXGXD sequence motif.[20] This fold is called a β-roll in the SCOP classification.
In pathology
[edit]Some proteins that are disordered or helical as monomers, such as amyloid β (see amyloid plaque) can form β-sheet-rich oligomeric structures associated with pathological states. The amyloid β protein's oligomeric form is implicated as a cause of Alzheimer's. Its structure has yet to be determined in full, but recent data suggest that it may resemble an unusual two-strand β-helix.[21]
The side chains from the amino acid residues found in a β-sheet structure may also be arranged such that many of the adjacent sidechains on one side of the sheet are hydrophobic, while many of those adjacent to each other on the alternate side of the sheet are polar or charged (hydrophilic),[22] which can be useful if the sheet is to form a boundary between polar/watery and nonpolar/greasy environments.
See also
[edit]References
[edit]- ^ "Animated GIF made by adapting a 3D model from Beta sheet - Proteopedia, Life in 3D". proteopedia.org.
- ^ Voet D, Voet JG (2004). Biochemistry (3rd ed.). Hoboken, NJ: Wiley. pp. 227–231. ISBN 0-471-19350-X.
- ^ Richardson JS, Richardson DC (March 2002). "Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation". Proceedings of the National Academy of Sciences of the United States of America. 99 (5): 2754–9. Bibcode:2002PNAS...99.2754R. doi:10.1073/pnas.052706099. PMC 122420. PMID 11880627.
- ^ Tertiary Protein Structure and Folds: section 4.3.2.1. From Principles of Protein Structure, Comparative Protein Modelling, and Visualisation
- ^ Hutchinson EG, Thornton JM (April 1993). "The Greek key motif: extraction, classification and analysis". Protein Engineering. 6 (3): 233–45. doi:10.1093/protein/6.3.233. PMID 8506258.
- ^ See sections II B and III C, D in Richardson JS (1981). "The Anatomy and Taxonomy of Protein Structure". Anatomy and Taxonomy of Protein Structures. Vol. 34. pp. 167–339. doi:10.1016/s0065-3233(08)60520-3. ISBN 0-12-034234-0. PMID 7020376.
{{cite book}}:|journal=ignored (help) - ^ a b Makabe K, McElheny D, Tereshko V, Hilyard A, Gawlak G, Yan S, et al. (November 2006). "Atomic structures of peptide self-assembly mimics". Proceedings of the National Academy of Sciences of the United States of America. 103 (47): 17753–8. Bibcode:2006PNAS..10317753M. doi:10.1073/pnas.0606690103. PMC 1693819. PMID 17093048.
- ^ "SCOP: Fold: WW domain-like". Archived from the original on 2012-02-04. Retrieved 2007-06-01.
- ^ "PPS '96 – Super Secondary Structure". Archived from the original on 2016-12-28. Retrieved 2007-05-31.
- ^ Biancalana M, Makabe K, Koide S (February 2010). "Minimalist design of water-soluble cross-beta architecture". Proceedings of the National Academy of Sciences of the United States of America. 107 (8): 3469–74. Bibcode:2010PNAS..107.3469B. doi:10.1073/pnas.0912654107. PMC 2840449. PMID 20133689.
- ^ Xu, Qingping; Biancalana, Matthew; Grant, Joanna C.; Chiu, Hsiu-Ju; Jaroszewski, Lukasz; Knuth, Mark W.; Lesley, Scott A.; Godzik, Adam; Elsliger, Marc-André; Deacon, Ashley M.; Wilson, Ian A. (September 2019). "Structures of single-layer β-sheet proteins evolved from β-hairpin repeats". Protein Science. 28 (9): 1676–1689. doi:10.1002/pro.3683. ISSN 1469-896X. PMC 6699103. PMID 31306512.
- ^ Hutchinson EG, Thornton JM (February 1996). "PROMOTIF--a program to identify and analyze structural motifs in proteins". Protein Science. 5 (2): 212–20. doi:10.1002/pro.5560050204. PMC 2143354. PMID 8745398.
- ^ a b Hutchinson EG, Thornton JM (1990). "HERA--a program to draw schematic diagrams of protein secondary structures". Proteins. 8 (3): 203–12. doi:10.1002/prot.340080303. PMID 2281084. S2CID 28921557.
- ^ Hubbard TJ, Murzin AG, Brenner SE, Chothia C (January 1997). "SCOP: a structural classification of proteins database". Nucleic Acids Research. 25 (1): 236–9. doi:10.1093/nar/25.1.236. PMC 146380. PMID 9016544.
- ^ Fox NK, Brenner SE, Chandonia JM (January 2014). "SCOPe: Structural Classification of Proteins--extended, integrating SCOP and ASTRAL data and classification of new structures". Nucleic Acids Research. 42 (Database issue): D304-9. doi:10.1093/nar/gkt1240. PMC 3965108. PMID 24304899.
- ^ Painter PC, Mosher LE, Rhoads C (July 1982). "Low-frequency modes in the Raman spectra of proteins". Biopolymers. 21 (7): 1469–72. doi:10.1002/bip.360210715. PMID 7115900.
- ^ Chou KC (August 1985). "Low-frequency motions in protein molecules. Beta-sheet and beta-barrel". Biophysical Journal. 48 (2): 289–97. Bibcode:1985BpJ....48..289C. doi:10.1016/S0006-3495(85)83782-6. PMC 1329320. PMID 4052563.
- ^ Liou YC, Tocilj A, Davies PL, Jia Z (July 2000). "Mimicry of ice structure by surface hydroxyls and water of a beta-helix antifreeze protein". Nature. 406 (6793): 322–4. Bibcode:2000Natur.406..322L. doi:10.1038/35018604. PMID 10917536. S2CID 4385352.
- ^ Branden C, Tooze J (1999). Introduction to Protein Structure. New York: Garland. pp. 20–32. ISBN 0-8153-2305-0.
- ^ Baumann U, Wu S, Flaherty KM, McKay DB (September 1993). "Three-dimensional structure of the alkaline protease of Pseudomonas aeruginosa: a two-domain protein with a calcium binding parallel beta roll motif". The EMBO Journal. 12 (9): 3357–64. doi:10.1002/j.1460-2075.1993.tb06009.x. PMC 413609. PMID 8253063.
- ^ Nelson R, Sawaya MR, Balbirnie M, Madsen AØ, Riekel C, Grothe R, Eisenberg D (June 2005). "Structure of the cross-beta pine of amyloid-like fibrils". Nature. 435 (7043): 773–8. Bibcode:2005Natur.435..773N. doi:10.1038/nature03680. PMC 1479801. PMID 15944695.
- ^ Zhang S, Holmes T, Lockshin C, Rich A (April 1993). "Spontaneous assembly of a self-complementary oligopeptide to form a stable macroscopic membrane". Proceedings of the National Academy of Sciences of the United States of America. 90 (8): 3334–8. Bibcode:1993PNAS...90.3334Z. doi:10.1073/pnas.90.8.3334. PMC 46294. PMID 7682699.
Further reading
[edit]- Cooper J (31 May 1996). "Super Secondary Structure - Part II". Principles of Protein Structure Using the Internet. Archived from the original on 28 December 2016. Retrieved 25 May 2007.
- "Open-sided Beta-meander". Structural Classification of Proteins (SCOP). 20 October 2006. Archived from the original on 4 February 2012. Retrieved 31 May 2007.