Recent from talks
Factor X
Knowledge base stats:
Talk channels stats:
Members stats:
Factor X
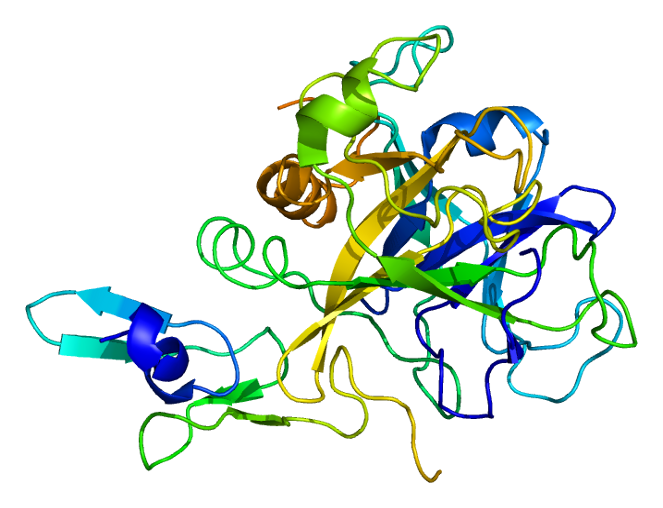
Coagulation factor X (EC 3.4.21.6), or Stuart factor, is an enzyme of the coagulation cascade, encoded in humans by F10 gene. It is a serine endopeptidase (protease group S1, PA clan). Factor X is synthesized in the liver and requires vitamin K for its synthesis.
Factor X is activated, by hydrolysis, into factor Xa by both factor IX with its cofactor, factor VIII in a complex known as intrinsic pathway; and factor VII with its cofactor, tissue factor in a complex known as extrinsic pathway. It is therefore the first member of the final common pathway or thrombin pathway.
It acts by cleaving prothrombin in two places (an Arg-Thr and then an Arg-Ile bond), which yields the active thrombin. This process is optimized when factor Xa is complexed with activated co-factor V in the prothrombinase complex.
Factor Xa is inactivated by protein Z-dependent protease inhibitor (ZPI), a serine protease inhibitor (serpin). The affinity of this protein for factor Xa is increased 1000-fold by the presence of protein Z, while it does not require protein Z for inactivation of factor XI. Defects in protein Z lead to increased factor Xa activity and a propensity for thrombosis. The half life of factor X is 40–45 hours.
The first crystal structure of human factor Xa was deposited in May 1993. To date, 191 crystal structures of factor Xa with various inhibitors have been deposited in the protein data bank. The active site of factor Xa is divided into four subpockets as S1, S2, S3 and S4. The S1 subpocket determines the major component of selectivity and binding. The S2 sub-pocket is small, shallow and not well defined. It merges with the S4 subpocket. The S3 sub-pocket is located on the rim of the S1 pocket and is quite exposed to solvent. The S4 sub-pocket has three ligand binding domains: the "hydrophobic box", the "cationic hole" and the water site. Factor Xa inhibitors generally bind in an L-shaped conformation, where one group of the ligand occupies the anionic S1 pocket lined by residues Asp189, Ser195, and Tyr228, and another group of the ligand occupies the aromatic S4 pocket lined by residues Tyr99, Phe174, and Trp215. Typically, a fairly rigid linker group bridges these two interaction sites.
The human factor X gene is located on chromosome 13 (13q34).
Inborn deficiency of factor X is very rare (1:1,000,000), and may present with epistaxis (nosebleeds), hemarthrosis (bleeding into joints) and gastrointestinal blood loss. Apart from congenital deficiency, low factor X levels may occur occasionally in a number of disease states. For example, factor X deficiency may be seen in amyloidosis, where factor X is adsorbed to the amyloid fibrils in the vasculature.
Deficiency of vitamin K or antagonism by warfarin (or similar medication) leads to the production of an inactive factor X. In warfarin therapy, this is desirable to prevent thrombosis. As of late 2007, four out of five emerging anti-coagulation therapeutics targeted this enzyme.
Hub AI
Factor X AI simulator
(@Factor X_simulator)
Factor X
Coagulation factor X (EC 3.4.21.6), or Stuart factor, is an enzyme of the coagulation cascade, encoded in humans by F10 gene. It is a serine endopeptidase (protease group S1, PA clan). Factor X is synthesized in the liver and requires vitamin K for its synthesis.
Factor X is activated, by hydrolysis, into factor Xa by both factor IX with its cofactor, factor VIII in a complex known as intrinsic pathway; and factor VII with its cofactor, tissue factor in a complex known as extrinsic pathway. It is therefore the first member of the final common pathway or thrombin pathway.
It acts by cleaving prothrombin in two places (an Arg-Thr and then an Arg-Ile bond), which yields the active thrombin. This process is optimized when factor Xa is complexed with activated co-factor V in the prothrombinase complex.
Factor Xa is inactivated by protein Z-dependent protease inhibitor (ZPI), a serine protease inhibitor (serpin). The affinity of this protein for factor Xa is increased 1000-fold by the presence of protein Z, while it does not require protein Z for inactivation of factor XI. Defects in protein Z lead to increased factor Xa activity and a propensity for thrombosis. The half life of factor X is 40–45 hours.
The first crystal structure of human factor Xa was deposited in May 1993. To date, 191 crystal structures of factor Xa with various inhibitors have been deposited in the protein data bank. The active site of factor Xa is divided into four subpockets as S1, S2, S3 and S4. The S1 subpocket determines the major component of selectivity and binding. The S2 sub-pocket is small, shallow and not well defined. It merges with the S4 subpocket. The S3 sub-pocket is located on the rim of the S1 pocket and is quite exposed to solvent. The S4 sub-pocket has three ligand binding domains: the "hydrophobic box", the "cationic hole" and the water site. Factor Xa inhibitors generally bind in an L-shaped conformation, where one group of the ligand occupies the anionic S1 pocket lined by residues Asp189, Ser195, and Tyr228, and another group of the ligand occupies the aromatic S4 pocket lined by residues Tyr99, Phe174, and Trp215. Typically, a fairly rigid linker group bridges these two interaction sites.
The human factor X gene is located on chromosome 13 (13q34).
Inborn deficiency of factor X is very rare (1:1,000,000), and may present with epistaxis (nosebleeds), hemarthrosis (bleeding into joints) and gastrointestinal blood loss. Apart from congenital deficiency, low factor X levels may occur occasionally in a number of disease states. For example, factor X deficiency may be seen in amyloidosis, where factor X is adsorbed to the amyloid fibrils in the vasculature.
Deficiency of vitamin K or antagonism by warfarin (or similar medication) leads to the production of an inactive factor X. In warfarin therapy, this is desirable to prevent thrombosis. As of late 2007, four out of five emerging anti-coagulation therapeutics targeted this enzyme.
Recent media