
Community hub
Recent from talks
Contribute something
Nothing was collected or created yet.
Fibrinolysis
View on WikipediaFibrinolysis is a process that prevents blood clots from growing and becoming problematic.[1] Primary fibrinolysis is a normal body process, while secondary fibrinolysis is the breakdown of clots due to a medicine, a medical disorder, or some other cause.[2]
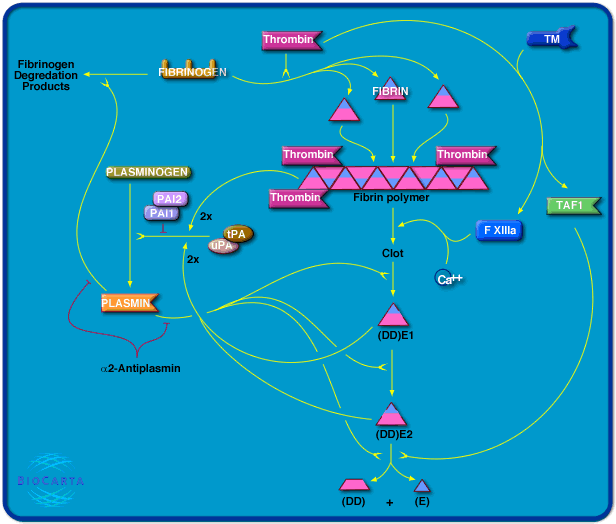
In fibrinolysis, a fibrin clot, the product of coagulation, is broken down.[3] Its main enzyme plasmin cuts the fibrin mesh at various places, leading to the production of circulating fragments that are cleared by other proteases or by the kidney and liver.
Physiology
[edit]
Plasmin is produced in an inactive form, plasminogen, in the liver. Although plasminogen cannot cleave fibrin, it still has an affinity for it, and is incorporated into the clot when it is formed.
Tissue plasminogen activator (t-PA)[4] and urokinase are the agents that convert plasminogen to the active plasmin, thus allowing fibrinolysis to occur. t-PA is released into the blood slowly by the damaged endothelium of the blood vessels, such that, after several days (when the bleeding has stopped), the clot is broken down. This occurs because plasminogen became entrapped within the clot when it formed; as it is slowly activated, it breaks down the fibrin mesh. t-PA and urokinase are themselves inhibited by plasminogen activator inhibitor-1 and plasminogen activator inhibitor-2 (PAI-1 and PAI-2). In contrast, plasminogen further stimulates plasmin generation by producing more active forms of both tissue plasminogen activator (tPA) and urokinase.
α2-Antiplasmin and α2-macroglobulin inactivate plasmin. Plasmin activity is also reduced by thrombin-activatable fibrinolysis inhibitor (TAFI), which modifies fibrin to make it more resistant to the tPA-mediated plasminogen.
Measurement
[edit]Plasmin breaks down fibrin into soluble parts called fibrin degradation products (FDPs). FDPs compete with thrombin, and thus slow down clot formation by preventing the conversion of fibrinogen to fibrin. This effect can be seen in the thrombin clotting time (TCT) test, which is prolonged in a person that has active fibrinolysis.
Antibody-antigen technology can measure FDPs and a specific FDP, the D-dimer. This is more specific than the TCT, and confirms that fibrinolysis has occurred. It is therefore used to indicate deep-vein thrombosis, pulmonary embolism, DIC, and efficacy of treatment in acute myocardial infarction. Alternatively, a more rapid detection of fibrinolytic activity, especially hyperfibrinolysis, is possible with thromboelastometry (TEM) in whole blood, even in patients on heparin. In this assay, increased fibrinolysis is assessed by comparing the TEM profile in the absence or presence of the fibrinolysis inhibitor aprotinin. Clinically, the TEM is useful for near real-time measurement of activated fibrinolysis for at-risk patients, such as those experiencing significant blood loss during surgery.[5]
Testing of overall fibrinolysis can be measured by a euglobulin lysis time (ELT) assay. The ELT measures fibrinolysis by clotting the euglobulin fraction (primarily the fibrinolytic factors fibrinogen, PAI-1, tPA, α2-antiplasmin, and plasminogen) from plasma and then observing the time required for clot dissolution. A shortened lysis time indicates a hyperfibrinolytic state and bleeding risk. Such results can be seen in peoples with liver disease, PAI-1 deficiency or α2-antiplasmin deficiency. Similar results are also seen after administration of desmopressin or after severe stress.[6]
Role in disease
[edit]Few congenital disorders of the fibrinolytic system have been documented. Nevertheless, excess levels of PAI and α2-antiplasmin have been implicated in metabolic syndrome and various other disease states.
However, acquired disturbance of fibrinolysis (hyperfibrinolysis), is not uncommon. Many trauma patients have an overwhelming activation of tissue factor and thus massive hyperfibrinolysis.[7] Hyperfibrinolysis may occur in other disease states. It could lead to massive bleeding if not diagnosed and treated early enough.
The fibrinolytic system is closely linked to control of inflammation, and plays a role in disease states associated with inflammation. Plasmin, in addition to lysing fibrin clots, also cleaves the complement system component C3, and fibrin degradation products have some vascular permeability inducing effects.
Pharmacology
[edit]In a process called thrombolysis (the breakdown of a thrombus), fibrinolytic drugs are used. They are given following a heart attack to dissolve the thrombus blocking the coronary artery; experimentally after a stroke to allow blood flow back to the affected part of the brain; and in the event of pulmonary embolism.[8]
Thrombolysis refers to the dissolution of the thrombus due to various agents while fibrinolysis refers specifically to the agents causing fibrin breakdown in the clot.
Antifibrinolytics, such as aminocaproic acid (ε-aminocaproic acid) and tranexamic acid are used as inhibitors of fibrinolysis. Their application may be beneficial in patients with hyperfibrinolysis because they arrest bleeding rapidly if the other components of the haemostatic system are not severely affected.[9] This may help to avoid the use of blood products such as fresh frozen plasma with its associated risks of infections or anaphylactic reactions.
Fibrinolytic enzymes
[edit]References
[edit]- ^ Mckenzie, Samuel (2019-03-27). "What is Fibrinolysis?". News-Medical. Retrieved 2024-05-31.
- ^ Dugdale D. "Fibrinolysis - primary or secondary". MedlinePlus. Retrieved August 7, 2011.
- ^ Cesarman-Maus G, Hajjar KA (May 2005). "Molecular mechanisms of fibrinolysis". British Journal of Haematology. 129 (3): 307–21. doi:10.1111/j.1365-2141.2005.05444.x. PMID 15842654.
- ^ Cotran RS, Kumar V, Collins T, Robbins SL (1999). Robbins pathologic basis of disease. Philadelphia: Saunders. ISBN 0-7216-7335-X. OCLC 39465455.
- ^ Levrat A, Gros A, Rugeri L, Inaba K, Floccard B, Negrier C, David JS (June 2008). "Evaluation of rotation thrombelastography for the diagnosis of hyperfibrinolysis in trauma patients". Br J Anaesth. 100 (6): 792–7. doi:10.1093/bja/aen083. PMID 18440953.
- ^ Goodnight SH, Hathaway WE (2001). Disorders of hemostasis and thrombosis: a clinical guide. New York: McGraw-Hill, Medical Pub. Division. ISBN 0-07-134834-4. OCLC 45485184.
- ^ Tieu BH, Holcomb JB, Schreiber MA (May 2007). "Coagulopathy: its pathophysiology and treatment in the injured patient". World J Surg. 31 (5): 1055–64. doi:10.1007/s00268-006-0653-9. PMID 17426904.
- ^ Patel N, Patel NJ, Agnihotri K, Panaich SS, Thakkar B, Patel A, et al. (December 2015). "Utilization of catheter-directed thrombolysis in pulmonary embolism and outcome difference between systemic thrombolysis and catheter-directed thrombolysis". Catheter Cardiovasc Interv. 86 (7): 1219–27. doi:10.1002/ccd.26108. PMID 26308961.
- ^ Levy JH, Koster A, Quinones QJ, Milling TJ, Key NS (March 2018). "Antifibrinolytic Therapy and Perioperative Considerations". Anesthesiology. 128 (3): 657–670. doi:10.1097/ALN.0000000000001997. PMC 5811331. PMID 29200009.
External links
[edit]- Human Gene Set: BIOCARTA_FIBRINOLYSIS_PATHWAY, Gene Set Enrichment Analysis, UC San Diego and Broad Institute
- Fibrinolysis Pathway chart, BioCarta (image linked from above page)
{kind=link}