Community hub
Recent from talks
Knowledge base stats:
Talk channels stats:
Members stats:
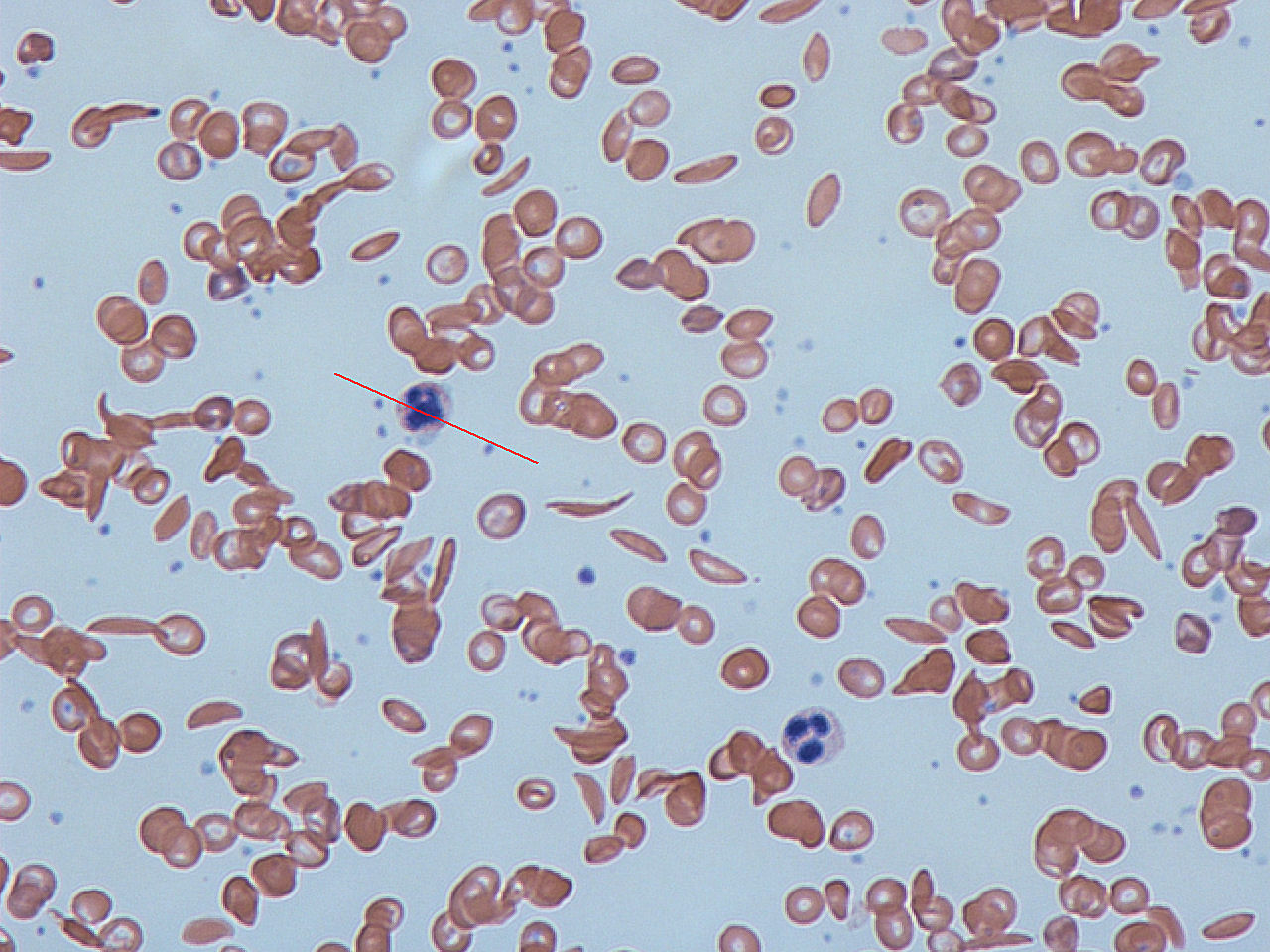
Hemoglobinopathy
Hemoglobinopathy is the medical term for a group of inherited blood disorders involving the hemoglobin, the major protein of red blood cells. They are generally single-gene disorders and, in most cases, they are inherited as autosomal recessive traits.
There are two main groups: abnormal structural hemoglobin variants caused by mutations in the hemoglobin genes, and the thalassemias, which are caused by an underproduction of otherwise normal hemoglobin molecules. The main structural hemoglobin variants are HbS, HbE and HbC. The main types of thalassemia are alpha-thalassemia and beta thalassemia.
Hemoglobin is a protein containing iron that facilitates the transportation of oxygen in red blood cells. Hemoglobin in the blood carries oxygen from the lungs to the other tissues of the body, where it releases the oxygen to enable aerobic respiration which powers the metabolism. Normal levels of hemoglobin vary according to sex and age in the range 9.5 to 17.2 grams of hemoglobin in every deciliter of blood.
Hemoglobin also transports other gases. It carries off some of the body's respiratory carbon dioxide (about 20–25% of the total) as carbaminohemoglobin, in which CO2 binds to the heme protein. The molecule also carries the important regulatory molecule nitric oxide bound to a thiol group in the globin protein, releasing it at the same time as oxygen.
Normal human hemoglobins are tetrameric proteins composed of two pairs of globin chains, each of which contains one alpha-like (α) globin and one beta-like (β) globin. Each globin chain is associated with an iron-containing heme moiety. Throughout life, the synthesis of the α and the β chains is balanced so that their ratio is relatively constant and there is no excess of either type.
The specific α and β chains that are incorporated into Hb are highly regulated during development:
Hemoglobin structural variants manifest a change in the structure of the Hb molecule. The majority of hemoglobin variants do not cause disease and are most commonly discovered either incidentally or through newborn screening. Hb variants can usually be detected by protein-based assay methods such as electrophoresis, isoelectric focusing, or high-performance liquid chromatography. Diagnosis is commonly confirmed by DNA sequencing.
The hemoglobin structural variants can be broadly classified as follows:
Hub AI
Hemoglobinopathy AI simulator
(@Hemoglobinopathy_simulator)
Hemoglobinopathy
Hemoglobinopathy is the medical term for a group of inherited blood disorders involving the hemoglobin, the major protein of red blood cells. They are generally single-gene disorders and, in most cases, they are inherited as autosomal recessive traits.
There are two main groups: abnormal structural hemoglobin variants caused by mutations in the hemoglobin genes, and the thalassemias, which are caused by an underproduction of otherwise normal hemoglobin molecules. The main structural hemoglobin variants are HbS, HbE and HbC. The main types of thalassemia are alpha-thalassemia and beta thalassemia.
Hemoglobin is a protein containing iron that facilitates the transportation of oxygen in red blood cells. Hemoglobin in the blood carries oxygen from the lungs to the other tissues of the body, where it releases the oxygen to enable aerobic respiration which powers the metabolism. Normal levels of hemoglobin vary according to sex and age in the range 9.5 to 17.2 grams of hemoglobin in every deciliter of blood.
Hemoglobin also transports other gases. It carries off some of the body's respiratory carbon dioxide (about 20–25% of the total) as carbaminohemoglobin, in which CO2 binds to the heme protein. The molecule also carries the important regulatory molecule nitric oxide bound to a thiol group in the globin protein, releasing it at the same time as oxygen.
Normal human hemoglobins are tetrameric proteins composed of two pairs of globin chains, each of which contains one alpha-like (α) globin and one beta-like (β) globin. Each globin chain is associated with an iron-containing heme moiety. Throughout life, the synthesis of the α and the β chains is balanced so that their ratio is relatively constant and there is no excess of either type.
The specific α and β chains that are incorporated into Hb are highly regulated during development:
Hemoglobin structural variants manifest a change in the structure of the Hb molecule. The majority of hemoglobin variants do not cause disease and are most commonly discovered either incidentally or through newborn screening. Hb variants can usually be detected by protein-based assay methods such as electrophoresis, isoelectric focusing, or high-performance liquid chromatography. Diagnosis is commonly confirmed by DNA sequencing.
The hemoglobin structural variants can be broadly classified as follows: