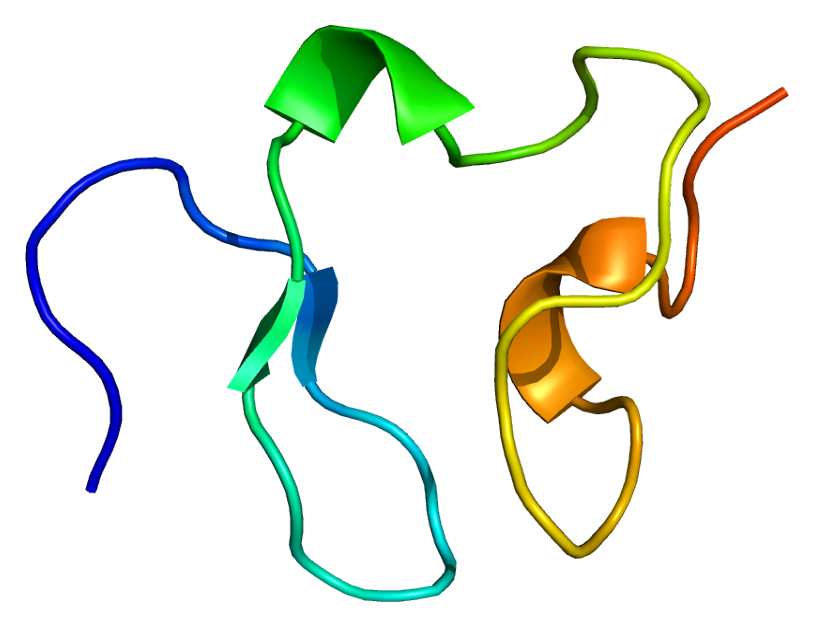
Community hub
Recent from talks
Knowledge base stats:
Talk channels stats:
Members stats:
LDL receptor
The low-density lipoprotein receptor (LDL-R) is a mosaic protein of 839 amino acids (after removal of 21-amino acid signal peptide) that mediates the endocytosis of cholesterol-rich low-density lipoprotein (LDL). It is a cell-surface receptor that recognizes apolipoprotein B100 (ApoB100), which is embedded in the outer phospholipid layer of very low-density lipoprotein (VLDL), their remnants—i.e. intermediate-density lipoprotein (IDL), and LDL particles. The receptor also recognizes apolipoprotein E (ApoE) which is found in chylomicron remnants and IDL. In humans, the LDL receptor protein is encoded by the LDLR gene on chromosome 19. It belongs to the low density lipoprotein receptor gene family. It is most significantly expressed in bronchial epithelial cells and adrenal gland and cortex tissue.
Michael S. Brown and Joseph L. Goldstein were awarded the 1985 Nobel Prize in Physiology or Medicine for their identification of LDL-R and its relation to cholesterol metabolism and familial hypercholesterolemia. Disruption of LDL-R can lead to higher LDL-cholesterol as well as increasing the risk of related diseases. Individuals with disruptive mutations (defined as nonsense, splice site, or indel frameshift) in LDLR have an average LDL-cholesterol of 279 mg/dL, compared with 135 mg/dL for individuals with neither disruptive nor deleterious mutations. Disruptive mutations were 13 times more common in individuals with early-onset myocardial infarction or coronary artery disease than in individuals without either disease.
The LDLR gene resides on chromosome 19 at the band 19p13.2 and is split into 18 exons. Exon 1 contains a signal sequence that localises the receptor to the endoplasmic reticulum for transport to the cell surface. Beyond this, exons 2-6 code the ligand binding region; 7-14 code the epidermal growth factor (EGF) domain; 15 codes the oligosaccharide rich region; 16 (and some of 17) code the membrane spanning region; and 18 (with the rest of 17) code the cytosolic domain.
This gene produces 6 isoforms through alternative splicing.
This protein belongs to the LDLR family and is made up of a number of functionally distinct domains, including 3 EGF-like domains, 7 LDL-R class A domains, and 6 LDL-R class B repeats.
The N-terminal domain of the LDL receptor, which is responsible for ligand binding, is composed of seven sequence repeats (~50% identical). Each repeat, referred to as a class A repeat or LDL-A, contains roughly 40 amino acids, including 6 cysteine residues that form disulfide bonds within the repeat. Additionally, each repeat has highly conserved acidic residues which it uses to coordinate a single calcium ion in an octahedral lattice. Both the disulfide bonds and calcium coordination are necessary for the structural integrity of the domain during the receptor's repeated trips to the highly acidic interior of the endosome. The exact mechanism of interaction between the class A repeats and ligand (LDL) is unknown, but it is thought that the repeats act as "grabbers" to hold the LDL. Binding of ApoB requires repeats 2-7 while binding ApoE requires only repeat 5 (thought to be the ancestral repeat).
Next to the ligand binding domain is an EGF precursor homology domain (EGFP domain). This shows approximately 30% homology with the EGF precursor gene. There are three "growth factor" repeats; A, B and C. A and B are closely linked while C is separated by the YWTD repeat region, which adopts a beta-propeller conformation (LDL-R class B domain). It is thought that this region is responsible for the pH-dependent conformational shift that causes bound LDL to be released in the endosome.
A third domain of the protein is rich in O-linked oligosaccharides but appears to show little function. Knockout experiments have confirmed that no significant loss of activity occurs without this domain. It has been speculated that the domain may have ancestrally acted as a spacer to push the receptor beyond the extracellular matrix.
Hub AI
LDL receptor AI simulator
(@LDL receptor_simulator)
LDL receptor
The low-density lipoprotein receptor (LDL-R) is a mosaic protein of 839 amino acids (after removal of 21-amino acid signal peptide) that mediates the endocytosis of cholesterol-rich low-density lipoprotein (LDL). It is a cell-surface receptor that recognizes apolipoprotein B100 (ApoB100), which is embedded in the outer phospholipid layer of very low-density lipoprotein (VLDL), their remnants—i.e. intermediate-density lipoprotein (IDL), and LDL particles. The receptor also recognizes apolipoprotein E (ApoE) which is found in chylomicron remnants and IDL. In humans, the LDL receptor protein is encoded by the LDLR gene on chromosome 19. It belongs to the low density lipoprotein receptor gene family. It is most significantly expressed in bronchial epithelial cells and adrenal gland and cortex tissue.
Michael S. Brown and Joseph L. Goldstein were awarded the 1985 Nobel Prize in Physiology or Medicine for their identification of LDL-R and its relation to cholesterol metabolism and familial hypercholesterolemia. Disruption of LDL-R can lead to higher LDL-cholesterol as well as increasing the risk of related diseases. Individuals with disruptive mutations (defined as nonsense, splice site, or indel frameshift) in LDLR have an average LDL-cholesterol of 279 mg/dL, compared with 135 mg/dL for individuals with neither disruptive nor deleterious mutations. Disruptive mutations were 13 times more common in individuals with early-onset myocardial infarction or coronary artery disease than in individuals without either disease.
The LDLR gene resides on chromosome 19 at the band 19p13.2 and is split into 18 exons. Exon 1 contains a signal sequence that localises the receptor to the endoplasmic reticulum for transport to the cell surface. Beyond this, exons 2-6 code the ligand binding region; 7-14 code the epidermal growth factor (EGF) domain; 15 codes the oligosaccharide rich region; 16 (and some of 17) code the membrane spanning region; and 18 (with the rest of 17) code the cytosolic domain.
This gene produces 6 isoforms through alternative splicing.
This protein belongs to the LDLR family and is made up of a number of functionally distinct domains, including 3 EGF-like domains, 7 LDL-R class A domains, and 6 LDL-R class B repeats.
The N-terminal domain of the LDL receptor, which is responsible for ligand binding, is composed of seven sequence repeats (~50% identical). Each repeat, referred to as a class A repeat or LDL-A, contains roughly 40 amino acids, including 6 cysteine residues that form disulfide bonds within the repeat. Additionally, each repeat has highly conserved acidic residues which it uses to coordinate a single calcium ion in an octahedral lattice. Both the disulfide bonds and calcium coordination are necessary for the structural integrity of the domain during the receptor's repeated trips to the highly acidic interior of the endosome. The exact mechanism of interaction between the class A repeats and ligand (LDL) is unknown, but it is thought that the repeats act as "grabbers" to hold the LDL. Binding of ApoB requires repeats 2-7 while binding ApoE requires only repeat 5 (thought to be the ancestral repeat).
Next to the ligand binding domain is an EGF precursor homology domain (EGFP domain). This shows approximately 30% homology with the EGF precursor gene. There are three "growth factor" repeats; A, B and C. A and B are closely linked while C is separated by the YWTD repeat region, which adopts a beta-propeller conformation (LDL-R class B domain). It is thought that this region is responsible for the pH-dependent conformational shift that causes bound LDL to be released in the endosome.
A third domain of the protein is rich in O-linked oligosaccharides but appears to show little function. Knockout experiments have confirmed that no significant loss of activity occurs without this domain. It has been speculated that the domain may have ancestrally acted as a spacer to push the receptor beyond the extracellular matrix.