
Community hub
Recent from talks
Contribute something
Nothing was collected or created yet.
Aryl halide
View on WikipediaIn organic chemistry, an aryl halide (also known as a haloarene) is an aromatic compound in which one or more hydrogen atoms directly bonded to an aromatic ring are replaced by a halide ion (such as fluorine F−, chlorine Cl−1,−3,−5, bromine Br−1, or iodine I−). Aryl halides are distinct from haloalkanes (alkyl halides) due to significant differences in their methods of preparation, chemical reactivity, and physical properties. The most common and important members of this class are aryl chlorides, but the group encompasses a wide range of derivatives with diverse applications in organic synthesis, pharmaceuticals, and materials science.
Classification according to halide
[edit]Aryl fluorides
[edit]Aryl fluorides are used as synthetic intermediates, e.g. for the preparation of pharmaceuticals, pesticides, and liquid crystals.[1] The conversion of diazonium salts is a well established route to aryl fluorides. Thus, anilines are precursors to aryl fluorides. In the classic Schiemann reaction, tetrafluoroborate is the fluoride donor:
- [C6H5N+2]BF−4 → C6H5F + N2 + BF3
In some cases, the fluoride salt is used:
- [C6H5N+2]F− → C6H5F + N2
Many commercial aryl fluorides are produced from aryl chlorides by the Halex process. The method is often used for aryl chlorides also bearing electron-withdrawing groups. Illustrative is the synthesis of 2-fluoronitrobenzene from 2-nitrochlorobenzene:[2]
- O2NC6H4Cl + KF → O2NC6H4F + KCl
Aryl chlorides
[edit]Aryl chlorides are the aryl halides produced on the largest scale commercially: 150,000 tons/y in the US alone (1994). Production levels are decreasing owing to environmental concerns. Chlorobenzenes are used mainly as solvents.[3]
Friedel-Crafts halogenation or "direct chlorination" is the main synthesis route. Lewis acids, e.g. iron(III) chloride, catalyze the reactions. The most abundantly produced aryl halide, chlorobenzene, is produced by this route:[4]
- C6H6 + Cl2 → C6H5Cl + HCl
Monochlorination of benzene is accompanied by formation of the dichlorobenzene derivatives.[3] Arenes with electron donating groups react with halogens even in the absence of Lewis acids. For example, phenols and anilines react quickly with chlorine and bromine water to give multihalogenated products. Many detailed laboratory procedures are available.[5] For alkylbenzene derivatives, e.g. toluene, the alkyl positions tend to be halogenated by free radical conditions, whereas ring halogenation is favored in the presence of Lewis acids.[6] The decolouration of bromine water by electron-rich arenes is used in the bromine test.

The oxychlorination of benzene has been well investigated, motivated by the avoidance of HCl as a coproduct in the direct halogenation:[3]
- 4 C6H6 + 4 HCl + O2 → 4 C6H5Cl + H2O
This technology is not widely used however.
The Gatterman reaction can also be used to convert diazonium salts to chlorobenzenes using copper-based reagents. Owing to high cost of diazonium salts, this method is reserved for specialty chlorides.
Aryl bromides
[edit]The main aryl bromides produced commercially are tetrabromophthalic anhydride, decabromodiphenyl ether, and tetrabromobisphenol-A. These materials are used as flame retardants. They are produced by direct bromination of phenols and aryl ethers. Phthalic anhydride is poorly reactive toward bromine, necessitating the use of acidic media.
The Gatterman reaction can also be used to convert diazonium salts to bromobenzenes using copper-based reagents. Owing to high cost of diazonium salts, this method is reserved for specialty bromides.
Aryl iodides
[edit]Synthetic aryl iodides are used as X-ray contrast agents, but otherwise these compounds are not produced on a large scale. Aryl iodides are "easy" substrates for many reactions such as cross-coupling reactions and conversion to Grignard reagents, but they are much more expensive than the lighter, less reactive aryl chlorides and bromides.
Aryl iodides can be prepared by treating diazonium salts with iodide salts.[7] Electron-rich arenes such as anilines and dimethoxy derivatives react directly with iodine.[8]
Aryl lithium and aryl Grignard reagents react with iodine to give the aryl halide:
- ArLi + I2 → ArI + LiI
This method is applicable to the preparation of all aryl halides. One limitation is that most, but not all,[9] aryl lithium and Grignard reagents are produced from aryl halides.
Classification according to aryl group
[edit]Halobenzenes and halobenzene derivatives
[edit]Although the term aryl halide includes halogenated derivatives of any aromatic compound, it commonly refers to halobenzenes, which are specifically halogenated derivatives of benzene. Groups of halobenzenes include fluorobenzenes, chlorobenzenes, bromobenzenes, and iodobenzenes, as well as mixed halobenzenes containing at least two different types of halogens bonded to the same benzene ring. There are also many halobenzene derivatives.
Halopyridines
[edit]Halopyridines are based on the aromatic compound pyridine.[10] This includes chloropyridines and bromopyridines. Chloropyridines are important intermediates to pharmaceuticals and agrochemicals.
Halogenated naphthalenes
[edit]Halogenated naphthalenes are based on naphthalene. Polychlorinated naphthalenes were used extensively from the 1930s to 1950s in cable and capacitor production, due to their insulating, hydrophobic, and flame retardant properties, but they have since been phased out for this use due to toxicity, environmental persistence, and introduction of new materials.[3]
Aryl halides in nature
[edit]The thyroid hormones triiodothyronine (T3) and thyroxine (T4) are aryl iodides. A tetraiodide, T4 is biosynthesised by electrophilic iodination of tyrosine derivative.[11] Synthetic T4 is one of the most heavily prescribed medicines in the U.S.[12]
Many chlorinated and brominated aromatic compounds are produced by marine organisms. Chloride and bromide ions in ocean waters are the source of the halogens. Various peroxidase enzymes (e.g., bromoperoxidase) catalyze the formation of these natural aryl chlorides and bromides. Numerous are derivatives of electron-rich rings found in tyrosine, tryptophan, and various pyrroles. Some of these natural aryl halides exhibit useful medicinal properties.[13][14]


Structural trends
[edit]The C-X distances for aryl halides follow the expected trend. These distances for fluorobenzene, chlorobenzene, bromobenzene, and methyl 4-iodobenzoate are 135.6(4), 173.90(23), 189.8(1), and 209.9 pm, respectively.[15]
Reactions
[edit]Substitution
[edit]Unlike typical alkyl halides, aryl halides typically do not participate in conventional substitution reactions. Aryl halides with electron-withdrawing groups in the ortho and para positions, can undergo SNAr reactions. For example, 2,4-dinitrochlorobenzene reacts in basic solution to give a phenol.
Unlike in most other substitution reactions, fluoride is the best leaving group, and iodide the worst.[16] A 2018 paper indicates that this situation may actually be rather common, occurring in systems that were previously assumed to proceed via SNAr mechanisms.[17]
Benzyne
[edit]When treated with strong base, some aryl halides often react via the intermediacy of benzynes. Benzyne is an intermediate in the reaction of chlorobenzene with strongly basic reagents such as potassium amide, even at −33 °C. It is also implicated in the conversion of chlorobenzene to phenol using sodium hydroxide, which requires high temperatures.[3][18]
Organometallic reagent formation
[edit]Aryl halides react with metals, generally lithium or magnesium, to give organometallic derivatives that function as sources of aryl anions. By the metal–halogen exchange reaction, aryl halides are converted to aryl lithium compounds. Illustrative is the preparation of phenyllithium from bromobenzene using n-butyllithium (n-BuLi):
- C6H5Br + BuLi → C6H5Li + BuBr
Direct formation of Grignard reagents, by adding the magnesium to the aryl halide in an ethereal solution, works well if the aromatic ring is not significantly deactivated by electron-withdrawing groups.

Other reactions
[edit]The halides can be displaced by strong nucleophiles via reactions involving radical anions. Alternatively aryl halides, especially the bromides and iodides, undergo oxidative addition, and thus are subject to Buchwald–Hartwig amination-type reactions.
Biodegradation
[edit]Rhodococcus phenolicus is a bacterium that degrades dichlorobenzene as sole carbon sources.[19]
Applications
[edit]
The aryl halides produced on the largest scale are chlorobenzene and the isomers of dichlorobenzene. One major but discontinued application was the use of chlorobenzene as a solvent for dispersing the herbicide Lasso. Overall, production of aryl chlorides (also naphthyl derivatives) has been declining since the 1980s, in part due to environmental concerns.[3] Triphenylphosphine is produced from chlorobenzene:
- 3 C6H5Cl + PCl3 + 6 Na → P(C6H5)3 + 6 NaCl
Some prominent herbicides are aryl chlorides.
- Aryl chloride-based herbicides
-

-

-
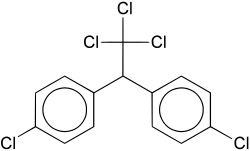
-
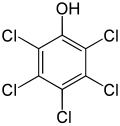




Several chlorobenzene derivatives are used as pigments and dyes.[20]
Aryl bromides are widely used as fire-retardants. A prominent member is tetrabromobisphenol-A, which is prepared by direct bromination of the diphenol.[21]
References
[edit]- ^ Shimizu, Masaki; Hiyama, Tamejiro (2005). "Modern Synthetic Methods for Fluorine-Substituted Target Molecules". Angewandte Chemie International Edition. 44 (2): 214–231. doi:10.1002/anie.200460441. PMID 15614922.
- ^ Siegemund, Günter; Schwertfeger, Werner; Feiring, Andrew; Smart, Bruce; Behr, Fred; Vogel, Herward; McKusick, Blaine (2002). "Fluorine Compounds, Organic". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a11_349. ISBN 978-3-527-30673-2..
- ^ a b c d e f Beck, U.; Löser, E. (2011). "Chlorinated Benzenes and Other Nucleus-Chlorinated Aromatic Hydrocarbons". Ullmann's Encyclopedia of Industrial Chemistry. doi:10.1002/14356007.o06_o03. ISBN 978-3527306732.
- ^ Peter Bernard, David De la Mare (1976). Electrophilic HalogenationReaction Pathways Involving Attack by Electrophilic Halogens on Unsaturated Compounds. Cambridge University Press. ISBN 9780521290142.
- ^ Atkinson, Edward R.; Murphy, Donald M.; Lufkin, James E. (1951). "dl-4,4′,6,6′-Tetrachlorodiphenic Acid". Organic Syntheses. 31: 96. doi:10.15227/orgsyn.031.0096.
- ^ Boyd, Robert W.; Morrison, Robert (1992). Organic chemistry. Englewood Cliffs, N.J: Prentice Hall. p. 947. ISBN 978-0-13-643669-0.
- ^ Lyday, Phyllis A.; Kaiho, Tatsuo (2015). "Iodine and Iodine Compounds". Ullmann's Encyclopedia of Industrial Chemistry. pp. 1–13. doi:10.1002/14356007.a14_381.pub2. ISBN 9783527306732.
- ^ Janssen, Donald E.; Wilson, C. V. (1956). "4-Iodoveratrole". Organic Syntheses. 36: 46. doi:10.15227/orgsyn.036.0046.
- ^ Snieckus, Victor (1990). "Directed ortho metalation. Tertiary amide and O-carbamate directors in synthetic strategies for polysubstituted aromatics". Chemical Reviews. 90 (6): 879–933. doi:10.1021/cr00104a001.
- ^ Siegemund, Günter; Schwertfeger, Werner; Feiring, Andrew; Smart, Bruce; Behr, Fred; Vogel, Herward; McKusick, Blaine (2000). "Pyridine and Pyridine Derivatives". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a22_399. ISBN 978-3-527-30673-2.
- ^ Mondal, Santanu; Raja, Karuppusamy; Schweizer, Ulrich; Mugesh, Govindasamy (2016). "Chemistry and Biology in the Biosynthesis and Action of Thyroid Hormones". Angewandte Chemie International Edition. 55 (27): 7606–7630. doi:10.1002/anie.201601116. PMID 27226395.
- ^ Brito, Juan P.; Ross, Joseph S.; El Kawkgi, Omar M.; Maraka, Spyridoula; Deng, Yihong; Shah, Nilay D.; Lipska, Kasia J. (2021). "Levothyroxine Use in the United States, 2008-2018". JAMA Internal Medicine. 181 (10): 1402–1405. doi:10.1001/jamainternmed.2021.2686. PMC 8218227. PMID 34152370.
- ^ Fujimori, Danica Galonić; Walsh, Christopher T. (2007). "What's New in Enzymatic Halogenations". Current Opinion in Chemical Biology. 11 (5): 553–60. doi:10.1016/j.cbpa.2007.08.002. PMC 2151916. PMID 17881282.
- ^ Gribble, Gordon W. (2004). "Natural Organohalogens: A New Frontier for Medicinal Agents?". Journal of Chemical Education. 81 (10): 1441. Bibcode:2004JChEd..81.1441G. doi:10.1021/ed081p1441.
- ^ Oberhammer, Heinz (2009). "The Structural Chemistry of Carbon-Halogen Bonds". PATai's Chemistry of Functional Groups. doi:10.1002/9780470682531.pat0002. ISBN 978-0-470-68253-1.
- ^ Ritter, Tobias; Hooker, Jacob M.; Neumann, Constanze N. (June 2016). "Concerted nucleophilic aromatic substitution with 19F− and 18F−". Nature. 534 (7607): 369–373. Bibcode:2016Natur.534..369N. doi:10.1038/nature17667. ISSN 1476-4687. PMC 4911285. PMID 27281221.
- ^ Jacobsen, Eric N.; Harrison A. Besser; Zeng, Yuwen; Kwan, Eugene E. (September 2018). "Concerted nucleophilic aromatic substitutions". Nature Chemistry. 10 (9): 917–923. Bibcode:2018NatCh..10..917K. doi:10.1038/s41557-018-0079-7. ISSN 1755-4349. PMC 6105541. PMID 30013193.
- ^ Smith, Michael B.; March, Jerry (2007), Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (6th ed.), New York: Wiley-Interscience, p. 870, ISBN 978-0-471-72091-1
- ^ Rehfuss, Marc; Urban, James (2005). "Rhodococcus phenolicus sp. nov., a novel bioprocessor isolated actinomycete with the ability to degrade chlorobenzene, dichlorobenzene and phenol as sole carbon sources". Systematic and Applied Microbiology. 28 (8): 695–701. doi:10.1016/j.syapm.2005.05.011. PMID 16261859.
- ^ K. Hunger. W. Herbst "Pigments, Organic" in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2012. doi:10.1002/14356007.a20_371
- ^ Ioffe, D.; Kampf, A. (2002). "Bromine, Organic Compounds". Kirk-Othmer Encyclopedia of Chemical Technology. doi:10.1002/0471238961.0218151325150606.a01. ISBN 978-0471238966.
Aryl halide
View on GrokipediaDefinition and Properties
Definition and Nomenclature
Aryl halides are organic compounds consisting of one or more halogen atoms directly bonded to a carbon atom of an aromatic ring system, such as benzene or other arenes.[1] The halogens involved are typically fluorine, chlorine, bromine, or iodine, with the general formula Ar-X, where Ar represents the aryl group and X the halogen.[1] Examples include bromobenzene (C6H5Br), chlorobenzene (C6H5Cl), and fluorobenzene (C6H5F).[1] In IUPAC nomenclature, aryl halides are classified as haloarenes, named by prefixing the halogen descriptor (fluoro-, chloro-, bromo-, or iodo-) to the parent arene name, with locants assigned to indicate positions when multiple substituents are present.[6] For benzene derivatives, monosubstituted compounds retain common names like chlorobenzene or bromobenzene, while polysubstituted ones use lowest locant rules or directional prefixes such as ortho- (1,2-), meta- (1,3-), or para- (1,4-).[6] In cases of fused or polycyclic arenes, the halogen is treated as a substituent on the systematic arene parent structure, following substitutive nomenclature principles.[7]Physical Properties
Aryl halides are typically colorless liquids or low-melting solids at room temperature, often possessing a characteristic aromatic odor.[8] Their physical properties, including boiling and melting points, are dominated by the non-polar aromatic ring, which enhances van der Waals interactions compared to alkyl halides of similar carbon count, leading to higher boiling points despite comparable molecular polarizability.[9] Boiling points increase with halogen atomic mass due to greater molecular weight and polarizability: fluorobenzene boils at 85 °C, chlorobenzene at 132 °C, bromobenzene at 156 °C, and iodobenzene at 188 °C.[10][8][11][12]| Halobenzene | Melting Point (°C) | Boiling Point (°C) | Density (g/cm³ at 20–25 °C) |
|---|---|---|---|
| Fluorobenzene | -44 | 85 | 1.025[10] |
| Chlorobenzene | -46 | 132 | 1.11[8] |
| Bromobenzene | -31 | 156 | 1.50[11] |
| Iodobenzene | -31 | 188 | 1.83[12] |
Structural and Reactivity Trends
Aryl halides feature a halogen atom bonded directly to an sp²-hybridized carbon of an aromatic ring, imparting distinct structural properties compared to their alkyl counterparts. The C–X bond (where X = F, Cl, Br, or I) is shorter and exhibits greater strength due to the partial double-bond character arising from resonance delocalization of the halogen's lone-pair electrons into the aromatic π-system.[14] This resonance stabilization hinders bond cleavage, rendering aryl halides far less susceptible to standard nucleophilic substitution mechanisms like SN1 or SN2, which are common for alkyl halides.[14] Additionally, the planar geometry of the sp² carbon precludes effective backside attack required for SN2, while phenyl-substituted carbocations are destabilized relative to alkyl analogs, disfavoring SN1 pathways.[15] Bond dissociation energies (BDEs) for aryl C–X bonds follow the trend C–F > C–Cl > C–Br > C–I, reflecting increasing ease of homolytic cleavage from fluorine to iodine; for example, the C–I BDE in iodobenzene is approximately 272 kJ/mol, lower than the ~500 kJ/mol for C–F.[16] This order influences reactivity in processes requiring C–X bond breaking, such as metal-catalyzed cross-couplings, where iodides and bromides are more reactive than chlorides or fluorides due to weaker bonds and better oxidative addition to transition metals.[17] However, in nucleophilic aromatic substitution (SNAr), the trend reverses for activated systems, with fluorides exhibiting highest reactivity owing to fluorine's electronegativity aiding stabilization of the negatively charged Meisenheimer intermediate. Substituent effects profoundly modulate reactivity, particularly for SNAr. Electron-withdrawing groups (e.g., nitro) at ortho or para positions to the halogen stabilize the Meisenheimer complex through delocalization, enhancing substitution rates by factors exceeding 108 relative to unactivated aryl halides.[14] Meta substituents exert minimal influence, as they cannot conjugate effectively with the reaction center.[18] Electron-donating groups deactivate the ring further. Unactivated aryl halides thus require alternative pathways, such as benzyne elimination-addition under strong basic conditions or catalytic methods, underscoring their overall inertness.[14]History
Early Isolation and Synthesis
Chlorobenzene, the simplest aryl chloride, was first synthesized in 1851 by reacting phenol with phosphorus pentachloride.[19] This approach leveraged the conversion of the phenolic hydroxyl group to chloride, yielding the compound in a manner analogous to alkyl halide formation from alcohols, though adapted for aromatic systems. The reaction proceeded via nucleophilic attack and elimination, producing chlorobenzene alongside phosphoryl chloride and HCl as byproducts. Bromobenzene was prepared similarly in the mid-19th century by treating phenol with phosphorus tribromide or pentabromide, reflecting the parallel development of halogenation techniques following bromine's isolation in 1826. Direct electrophilic aromatic substitution on benzene using halogens and metal catalysts, such as iron or antimony salts, emerged soon after for scalable synthesis; for instance, chlorination of benzene in the presence of ferric chloride afforded chlorobenzene via substitution, contrasting earlier addition products obtained without catalysts. These methods established aryl halides as accessible synthetic intermediates despite their relative inertness compared to alkyl halides.Key Milestones in Reactivity and Applications
In the mid-19th century, aryl halides were noted for their markedly lower reactivity compared to alkyl halides in nucleophilic substitution, owing to the sp²-hybridized carbon-halogen bond and lack of facile backside attack.[1] One early milestone was the Fittig reaction, reported around 1860, in which aryl halides such as bromobenzene reacted with sodium metal to form biaryls like biphenyl, or with alkyl halides in the related Wurtz-Fittig variant to yield alkyl-substituted arenes, demonstrating initial carbon-carbon coupling potential despite harsh conditions.[20] A pivotal advancement occurred in 1900 when Victor Grignard discovered the preparation of organomagnesium reagents by reacting magnesium with halides in ether, applicable to aryl halides like bromobenzene to form phenylmagnesium bromide, which facilitated nucleophilic additions to carbonyls and expanded synthetic utility in constructing complex carbon frameworks.[21] This was closely followed in 1901 by Fritz Ullmann's copper-mediated diaryl coupling of aryl iodides or bromides, enabling efficient biaryl formation under thermal conditions with copper powder, a reaction that became foundational for synthesizing pharmaceuticals, dyes, and materials despite requiring high temperatures.[22] Nucleophilic aromatic substitution (SNAr) reactivity, viable primarily with electron-withdrawing groups ortho or para to the halide, emerged as a key pathway by the early 20th century, with foundational studies dating to the 1870s and mechanistic insights from Meisenheimer complex isolation in 1902 confirming the addition-elimination sequence via stabilized anionic intermediates.[23] Applications expanded post-1930s, including aryl halide intermediates in herbicide synthesis like 2,4-dichlorophenoxyacetic acid (1940s) and insecticides such as DDT (commercialized 1942 from earlier 1874 synthesis), highlighting their role in agrochemicals despite environmental concerns later identified.[24] These developments underscored aryl halides' transition from inert solvents to versatile synthons in cross-coupling precursors.Natural Occurrence
Sources in Nature
Aryl halides occur naturally, albeit infrequently relative to aliphatic organohalogens, primarily as components of hormones, antibiotics, and microbial metabolites. In vertebrates, the thyroid gland synthesizes thyroxine (T4, or 3,5,3',5'-tetraiodo-L-thyronine), which incorporates four iodine atoms bonded directly to the aromatic rings of its diphenyl ether structure derived from tyrosine residues; this hormone regulates metabolism, growth, and development. Triiodothyronine (T3), a deiodinated analog with three iodine substituents on the aromatic framework, functions similarly as the more active form. These iodinated aryl structures arise via enzymatic iodination using dietary iodide in the thyroid follicular cells. Microbial sources include soil actinomycetes such as Amycolatopsis orientalis, which produce vancomycin, a glycopeptide containing two chlorine atoms attached to the aromatic rings of its heptapeptide aglycone core; these chlorides enhance binding to bacterial cell wall precursors, conferring activity against Gram-positive pathogens. Vancomycin's biosynthesis involves halogenase enzymes that install the aryl chlorides during nonribosomal peptide assembly. Other chlorinated aryl-containing antibiotics, such as those from streptomycetes, similarly feature halogenated aromatics for bioactivity.[25] Terrestrial fungi, particularly wood-decomposing basidiomycetes, generate chlorinated aromatic compounds like chlorophenols and related structures in forest soils, often at concentrations exceeding 100 μg/kg dry weight; these arise from fungal haloperoxidases utilizing chloride ions. Marine environments yield brominated aryl halides, though predominantly in complex secondary metabolites from algae and sponges rather than simple monomers; examples include bromophenols from red algae (Rhodophyta), biosynthesized via bromoperoxidases accessing abundant seawater bromide.[26] Such natural aryl bromides contribute to chemical defense against herbivores and microbes.[26] Overall, natural aryl halide production relies on halide availability and enzymatic halogenation, contrasting with the prevalence of synthetic aryl halides in industry.Biological Functions and Biosynthesis
Aryl halides occur in nature primarily as iodinated derivatives in vertebrate hormones and chlorinated or brominated components in microbial and marine natural products. Thyroid hormones thyroxine (T4) and 3,5,3'-triiodothyronine (T3) exemplify aryl iodides, with three or four iodine atoms substituting hydrogens on the phenolic rings of coupled diiodotyrosine units derived from tyrosine. These hormones are essential for regulating basal metabolic rate, thermogenesis, growth, and differentiation in mammals and other vertebrates.[27][28] Biosynthesis of T4 and T3 occurs in the thyroid gland, where dietary iodide is actively transported into follicular cells by the sodium-iodide symporter (NIS), achieving concentrations up to 20-40 times higher than plasma levels. Thyroid peroxidase (TPO) then oxidizes iodide using hydrogen peroxide to generate electrophilic iodine species, such as I+ or hypoiodous acid (HOI), which perform regioselective iodination on tyrosyl residues within the thyroglobulin protein matrix; mono- and diiodotyrosines subsequently couple to form T3 and T4, which are stored until proteolytic release.[27][28] In bacteria, aryl chlorides feature in glycopeptide antibiotics like vancomycin, produced by Amycolatopsis orientalis, where two chlorine atoms on β-hydroxytyrosine-derived aromatic rings rigidify the structure and enhance binding to D-Ala-D-Ala termini of peptidoglycan precursors, inhibiting cell wall synthesis in Gram-positive pathogens. The chlorines contribute to increased potency against resistant strains, with dechlorinated analogs showing reduced activity by factors of 10-100.[29][30] Vancomycin's chlorination occurs early in non-ribosomal peptide synthetase (NRPS) assembly, mediated by the flavin adenine dinucleotide (FAD)-dependent halogenase VhaA, which abstracts a hydrogen from the activated aromatic ring of a tyrosine precursor and facilitates chloride incorporation via hypochlorite-like intermediates; this timing, confirmed by trapping chlorinated heptapeptide intermediates, ensures halogenation before cyclization and glycosylation.[29] Brominated aryl halides appear in marine ecosystems, such as in the biosynthesis of Tyrian purple (6,6'-dibromoindigo) precursors from muricid gastropods, where bromoperoxidases catalyze regioselective bromination of indoxyl sulfate using bromide from seawater, yielding defensive pigments or metabolites with potential antimicrobial roles, though exact functions remain understudied.[32] Overall, biological halogenation of aromatic rings enhances molecular lipophilicity, receptor affinity, and stability, often via dedicated enzymes like flavin-dependent halogenases for precise C-H abstraction and halide insertion or vanadium haloperoxidases for broader electrophilic halogenation, contrasting abiotic processes and enabling tailored bioactivities in secondary metabolism.[33][34]Synthesis Methods
Classical Halogenation Approaches
Classical halogenation of aromatic compounds represents a foundational method for synthesizing aryl chlorides and bromides via electrophilic aromatic substitution. In this process, benzene reacts with chlorine or bromine gas in the presence of a Lewis acid catalyst, such as iron(III) chloride (FeCl₃) or aluminum chloride (AlCl₃), under anhydrous conditions at moderate temperatures (typically 0–50 °C). The reaction yields chlorobenzene or bromobenzene, respectively, along with hydrochloric or hydrobromic acid as byproducts.[35][36] To minimize polyhalogenation, an excess of the arene substrate is employed, though selective monohalogenation remains challenging without precise control.[37] The mechanism initiates with the coordination of the Lewis acid to the halogen molecule, polarizing it to generate an electrophilic species equivalent to X⁺ (where X = Cl or Br). This electrophile then attacks the electron-rich π-system of the aromatic ring, forming a resonance-stabilized arenium ion intermediate (also known as the Wheland intermediate or σ-complex). Subsequent deprotonation by the conjugate base (e.g., FeCl₄⁻) regenerates the aromatic system and releases the proton, completing the substitution.[38][39] The halogen substituents introduced are ortho-para directors but deactivators in further substitutions due to their electronegativity, influencing regioselectivity in polysubstituted products.[40] Bromination follows an analogous pathway but proceeds more readily than chlorination owing to the greater polarizability of Br₂, allowing milder conditions and often higher yields for monobromination. Iodination, however, requires activation of iodine with an oxidant like nitric acid or iodic acid, as I₂ alone lacks sufficient electrophilicity; the reaction typically yields aryl iodides in lower efficiency and is prone to over-oxidation. Direct fluorination is impractical under these classical conditions, as fluorine gas reacts violently with arenes, leading to substitution, addition, or ring disruption rather than clean aryl fluoride formation.[41][42] These methods are most effective for electron-rich or unsubstituted arenes, with deactivated substrates (e.g., nitrobenzene) showing poor reactivity due to insufficient electron density in the ring.[1]Diazonium-Based Preparations
Aryldiazonium salts, prepared by diazotization of primary arylamines with nitrous acid (typically generated from sodium nitrite and mineral acid at 0–5 °C), serve as key intermediates for synthesizing aryl chlorides, bromides, and iodides via copper-mediated displacements.[43] In the Sandmeyer reaction, the diazonium salt reacts with copper(I) chloride or bromide in aqueous solution to yield the corresponding aryl chloride or bromide, accompanied by nitrogen gas evolution; aryl iodides can be obtained similarly using potassium iodide, often without copper catalysis.[43][44] This method proceeds via a radical mechanism wherein copper(I) reduces the diazonium ion to an aryl radical, which abstracts a halogen atom from the copper(II) halide intermediate.[44][45] A variant, the Gatterman reaction, employs copper powder in concentrated hydrohalic acid (HCl or HBr) with the diazonium salt to produce aryl chlorides or bromides, offering an alternative to the Sandmeyer process for cases where soluble copper salts are less effective.[46][47] Unlike direct electrophilic halogenation, these diazonium routes enable halide introduction ortho or para to electron-donating groups that would otherwise deactivate the ring toward halogenation.[42] Yields typically range from 50–80%, limited by the thermal instability of diazonium salts, which necessitates in situ generation and low-temperature handling to prevent explosive decomposition.[45] For aryl fluorides, the Balz–Schiemann reaction involves precipitating the diazonium salt as its tetrafluoroborate (ArN₂⁺ BF₄⁻) with boron trifluoride or sodium tetrafluoroborate, followed by thermal decomposition at 100–200 °C to afford the aryl fluoride.[48] This proceeds through loss of nitrogen to form an aryl cation, which captures fluoride from the BF₄⁻ counterion, though competing rearrangements can reduce selectivity for ortho-substituted substrates.[48][49] Direct treatment of diazonium chlorides with aqueous HF yields fluorides in poor efficiency due to side reactions, making the Balz–Schiemann the standard despite modest yields (40–70%) and the need for anhydrous conditions.[48] These methods complement catalytic approaches by providing access to halides in electron-rich aromatics, though they are incompatible with acid-sensitive functional groups.[50]Contemporary Catalytic and Electrochemical Routes
Transition metal-catalyzed C-H halogenation has emerged as a key contemporary strategy for synthesizing aryl halides with improved regioselectivity and milder conditions compared to classical electrophilic aromatic substitution, which often suffers from over-halogenation. These methods typically employ directing groups, such as amides or ureas, to guide the catalyst to specific ortho positions, using N-halosuccinimides (e.g., NBS for bromination, NCS for chlorination) as halogen sources. For example, palladium(II) catalysts facilitate ortho-selective halogenation of anilides, yielding aryl bromides or chlorides in high yields under aerobic conditions at moderate temperatures.[51] Similarly, rhodium(III)-catalyzed C-H halogenation of 2-arylbenzothiazoles with NXS provides ortho-bromo or iodo derivatives with turnover numbers up to 80.[52] First-row transition metals offer economical alternatives, enabling site-selective halogenation of (hetero)arenes without precious metals. Iron-catalyzed chlorination of indoles using NCS achieves mono-substitution at the 3-position with yields exceeding 90%, while cobalt catalysts support bromination of pyridines via directing-group assistance.[53] Metal-free catalytic variants, such as carborane-based Lewis bases, promote electrophilic halogenation of electron-rich arenes with NXS, delivering monohalogenated products in up to 99% yield by stabilizing the halogenating agent and enhancing selectivity.[54] These approaches minimize waste and enable late-stage functionalization, though challenges persist for unactivated arenes lacking directing groups. Electrochemical methods provide oxidant-free routes to aryl halides by leveraging anodic generation of halogen equivalents from halide salts. Direct C-H halogenation of N-heteroarenes, such as indoles and pyrroles, occurs selectively at electron-rich positions using undivided cells with chloride or bromide electrolytes, affording 2- or 3-halo derivatives in 70-95% yields without catalysts.[55] For electron-rich arenes like phenols or anilines, anodic oxidation in the presence of halide ions facilitates regioselective chlorination or bromination, bypassing traditional oxidants and achieving current efficiencies above 80%.[56] These techniques align with green chemistry principles, utilizing electricity as the driving force, but scalability remains limited by electrode fouling in complex substrates.Chemical Reactions
Nucleophilic Aromatic Substitution
Nucleophilic aromatic substitution (SNAr) on aryl halides proceeds via an addition-elimination mechanism, distinct from the direct displacement seen in aliphatic systems, due to the sp² hybridization of the ipso carbon and resonance stabilization of the C–X bond that hinders backside attack.[57] Unactivated aryl halides exhibit negligible reactivity under typical conditions, as the transition state for nucleophilic addition would disrupt aromaticity without sufficient stabilization.[18] Reactivity requires activation by one or more electron-withdrawing groups (EWGs), such as nitro (–NO₂), cyano (–CN), or carbonyl moieties, positioned ortho or para to the halide; these groups stabilize the developing negative charge in the intermediate through resonance delocalization.[58] Ortho/para activation is more effective than meta, as the former allows direct conjugation with the addition site, lowering the activation energy for nucleophilic attack by up to several orders of magnitude.[59] Fluorine serves as the most labile halide in activated SNAr due to its electronegativity facilitating the addition step, with reactivity order F > Cl > Br > I observed experimentally for poly-nitro substituted systems.[18] The mechanism initiates with nucleophilic addition to the ipso carbon, forming a cyclohexadienyl anion known as the Meisenheimer complex, where the ring temporarily loses aromaticity and the negative charge is dispersed via resonance involving the EWG./Chapter_13._Addition-Elimination_Sequences/13.1:Nucleophilic_Addition-Elimination/13.1.2"Nucleophilic_Aromatic_Substitution") This intermediate, isolable in some cases under cryogenic conditions, then undergoes base-promoted elimination of the halide ion to restore aromaticity and yield the substitution product; the addition step is typically rate-determining for activated substrates.[60] Kinetic isotope effect studies confirm the stepwise nature, with primary KIE values near 1 for carbon-halogen bonds indicating no C–X cleavage in the rate-limiting step.[61] Common examples include the displacement of chloride in 1-chloro-2,4-dinitrobenzene by amines or alkoxides in polar aprotic solvents like DMF at elevated temperatures (e.g., 100–150°C), yielding aryl ethers or anilines with yields exceeding 80% under optimized conditions.[58] Similarly, 2,4,6-trinitrochlorobenzene reacts rapidly with hydroxide to form picric acid derivatives, demonstrating how multiple ortho/para nitro groups enhance rates by factors of 10⁴–10⁶ relative to mono-activated analogs.[18] While SNAr dominates for electron-deficient heterocycles like 2-chloropyridines, carbocyclic aryl halides require stringent activation to avoid competing elimination pathways.[60]Elimination Mechanisms Including Benzyne Formation
Aryl halides lacking electron-withdrawing ortho or para substituents resist direct nucleophilic aromatic substitution via the addition-elimination pathway and instead undergo an elimination-addition mechanism under forcing basic conditions, generating a highly reactive benzyne (dehydrobenzene) intermediate.[62][63] This process requires very strong bases, such as sodamide (NaNH₂) in liquid ammonia at elevated temperatures (typically around 100–150°C) or potassium amide (KNH₂), to initiate deprotonation at the ortho position relative to the halogen.[64][65] Iodides and bromides react more readily than chlorides or fluorides due to the better leaving group ability of I⁻ and Br⁻, with fluoride halides being particularly inert.[63] The elimination step begins with the strong base abstracting a proton from the carbon ortho to the halogen, forming a transient aryl anion; this is followed rapidly by expulsion of the halide ion, yielding benzyne, a strained species featuring a formal carbon-carbon triple bond within the aromatic ring (bond angles distorted to ~120° instead of 180° for sp hybridization).[62][63] This elimination is facilitated by the thermodynamic driving force of aromatization but is kinetically demanding, often requiring aprotic solvents like liquid ammonia to solvate the base effectively and minimize protonation side reactions.[66] Benzyne's electrophilicity arises from the electron-deficient triple bond, enabling rapid addition of nucleophiles such as amide (NH₂⁻), alkoxides, or hydride; the nucleophile bonds to one of the triple-bonded carbons, generating an aryl anion that is then protonated at the adjacent position to restore aromaticity.[67][65] In unsymmetrically substituted aryl halides, benzyne formation can occur from either ortho position, and subsequent nucleophilic addition yields regioisomeric products, often in comparable ratios (e.g., ~50:50 for ortho-methylchlorobenzene yielding o- and m-toluidine upon amination).[58][63] Regioselectivity in addition is influenced by inductive effects of substituents or nucleophile basicity, with electron-donating groups directing the anion-stabilizing protonation step away from themselves.[62] The existence of benzyne as an intermediate was confirmed in the 1950s through isotopic labeling experiments: J.D. Roberts and colleagues used ¹⁴C-labeled chlorobenzene derivatives, observing equivalent labeling in ortho and meta positions of the product aniline, inconsistent with direct substitution but supportive of symmetric benzyne addition; similar results from deuterium substitution in ortho positions yielded meta-deuterated products, ruling out simple proton abstraction without rearrangement.[58][62] Alternative elimination pathways are rare for simple aryl halides but include SRN1 mechanisms under irradiation with electron donors, involving radical anions rather than benzyne; however, these require specific initiators and do not predominate in standard basic conditions.[68] Benzyne generation has synthetic utility for aryne cycloadditions (e.g., Diels-Alder with dienes) but is limited by the intermediate's fleeting lifetime (~10⁻¹² s in solution) and competing polymerization or protonation side products.[66][63]Organometallic Derivative Formation
Aryl halides, particularly bromides and iodides, undergo oxidative addition with magnesium metal to form aryl Grignard reagents (ArMgX) under anhydrous conditions in solvents such as diethyl ether or tetrahydrofuran (THF).[69] [70] The reaction proceeds via a single-electron transfer mechanism, though aryl radical intermediates are not involved, unlike in some alkyl cases.[70] Reactivity decreases from iodide to bromide to chloride, with chlorides often requiring catalysts or activated magnesium for efficient conversion.[71] These reagents are highly reactive nucleophiles used in subsequent carbon-carbon bond formations. Organolithium compounds (ArLi) are commonly prepared from aryl halides via lithium-halogen exchange rather than direct metallation, especially for functionalized aryl bromides and iodides.[72] This involves treating the aryl halide with an alkyllithium reagent, such as n-butyllithium or tert-butyllithium, in THF at low temperatures (typically -78°C) to generate ArLi rapidly and selectively, displacing the halide with lithium from the alkyl group.[73] [74] The exchange is faster than direct insertion and avoids side reactions like elimination, making it preferable for sensitive substrates.[72] The mechanism involves a polar pathway with possible radical character, but proceeds without persistent radicals under standard conditions.[75] Other organometallic derivatives, such as organozinc compounds, can be formed from aryl Grignard or organolithium reagents via transmetalation, though direct insertion of zinc into aryl halides is less common and typically requires activation.[76] These derivatives enable milder reactivity profiles for applications like Negishi coupling, where direct Grignard or organolithium use might be incompatible.[76] Formation conditions must exclude protic impurities and oxygen to prevent decomposition, emphasizing the need for rigorous inert atmosphere techniques.[69]Cross-Coupling and Functionalization Reactions
Cross-coupling reactions represent a cornerstone of modern synthetic chemistry, utilizing aryl halides as electrophilic partners in transition metal-catalyzed processes to forge carbon-carbon and carbon-heteroatom bonds with high selectivity and efficiency. These transformations, pioneered in the late 20th century, rely on a general catalytic cycle involving oxidative addition of the C-X bond to a low-valent metal (typically palladium), transmetalation with a nucleophilic partner, and reductive elimination to yield the coupled product. Aryl iodides and bromides exhibit superior reactivity compared to chlorides due to weaker bond strengths, though ligand innovations have expanded scope to less reactive chlorides and pseudohalides like triflates.[77][78] The Suzuki-Miyaura coupling, first reported in 1979, couples aryl or vinyl halides with boronic acids or esters under palladium catalysis and basic conditions to produce biaryls, a motif prevalent in pharmaceuticals and agrochemicals. The reaction's mechanism proceeds via oxidative addition to form an arylpalladium(II) halide, transmetalation facilitated by base activation of the boron reagent, and reductive elimination; its popularity stems from boronic acids' commercial availability, air stability, and functional group tolerance. Variations include aqueous protocols and immobilizations for scalability, with turnover numbers exceeding 10^6 in optimized systems.[79][80] In the Mizoroki-Heck reaction, discovered independently by Tsutomu Mizoroki in 1971 and Richard F. Heck in 1972, aryl halides react with alkenes in the presence of palladium catalysts and bases to afford aryl-substituted alkenes via syn-β-hydride elimination, enabling stereoselective access to trans-stilbenes and cinnamates. The process favors electron-deficient alkenes like acrylates and exhibits regioselectivity influenced by ligand choice, with bidentate phosphines enhancing activity for aryl bromides. Recent adaptations include asymmetric variants using chiral ligands for enantioselective olefin functionalization.[81][82] For heteroatom couplings, the Buchwald-Hartwig amination, advanced in the 1990s by Stephen Buchwald and John Hartwig, employs palladium catalysts with bulky phosphine or N-heterocyclic carbene ligands to couple aryl halides with amines, yielding arylamines crucial for drug scaffolds. The reaction accommodates primary, secondary, and even ammonia equivalents, with mechanisms involving amine coordination post-oxidative addition and base-mediated deprotonation; electron-rich aryl chlorides now react under mild conditions due to ligand tuning. Extensions to C-O and C-S bond formation follow analogous pathways, broadening functionalization to ethers and thioethers.[83][84] Other notable cross-couplings include the Sonogashira reaction for aryl alkyne synthesis from terminal alkynes and aryl halides, and the Stille coupling with organostannanes, both palladium-mediated but requiring copper co-catalysis or toxic tin partners, respectively. Emerging ligand-free and nickel-catalyzed variants reduce costs and expand to unactivated substrates, while electroreductive and photoredox methods enable radical-mediated couplings under milder conditions, minimizing palladium reliance. These advances have facilitated over 90% of C-C bond formations in complex molecule total syntheses reported since 2010.[77][85]Applications and Industrial Uses
Materials and Polymers
Aryl halides function as essential monomers in the synthesis of conjugated polymers through transition metal-catalyzed cross-coupling reactions, including direct arylation polymerization (DAP), which couples aryl halides with aromatic C-H bonds to yield high molecular weight π-conjugated systems without requiring preformed organometallics.[86] These polymers, such as polythiophenes, polyfluorenes, and polycarbazoles, exhibit tunable electronic properties suited for applications in organic semiconductors.[87] Palladium- and nickel-catalyzed variants of DAP enable the polymerization of non-activated aryl halides with thiophene or arene units, producing materials for organic photovoltaics, light-emitting diodes, and thin-film transistors, with molecular weights often exceeding 20 kDa and polydispersity indices below 2.5.[88] For instance, Suzuki polycondensation of aryl dichlorides with aryl diboronic esters generates polyarylenes with high thermal stability (decomposition temperatures above 400°C) and mechanical strength, used in high-performance coatings and fibers.[89] In engineering plastics, dihaloarenes like p-dichlorobenzene undergo nucleophilic substitution with sodium sulfide to form polyphenylene sulfide (PPS), a semicrystalline thermoplastic with a glass transition temperature of 85–90°C and melting point of 280°C, valued for its chemical resistance and use in automotive and electronic components produced at scales exceeding 100,000 tons annually.[90] Aryl halides also serve as precursors for polycyclic aromatic hydrocarbons (PAHs) via iterative cross-couplings, incorporating into carbon-rich materials for advanced composites and graphene analogs.[91]Pharmaceuticals and Agrochemicals
Aryl halides serve as essential building blocks and active components in pharmaceutical synthesis, where their reactivity in cross-coupling reactions facilitates the construction of complex therapeutic molecules. The Buchwald-Hartwig C-N coupling of aryl halides with amines ranks among the top 20 most used reactions in medicinal chemistry, enabling the formation of arylamines prevalent in drugs targeting kinases and G-protein coupled receptors.[92] Specific pharmaceuticals incorporate aryl halides directly; for example, the antiarrhythmic amiodarone features diiodinated aryl rings that contribute to its efficacy in treating ventricular arrhythmias, while the ALK inhibitor crizotinib contains a 2,6-dichlorophenyl moiety critical for its antitumor activity.[93] In agrochemicals, aryl chlorides predominate in the structures of legacy and contemporary herbicides and pesticides, enhancing selectivity and potency through modulation of electronic properties and binding interactions. The herbicide 2,4-dichlorophenoxyacetic acid (2,4-D), bearing two chlorine substituents on its phenyl ring, acts as a synthetic auxin to selectively kill broadleaf weeds and has been in commercial use since the 1940s, with global production exceeding 50,000 tons annually as of recent estimates.[94] Similarly, dicamba, with its 3,6-dichlorinated benzoic acid core, targets auxin pathways in susceptible plants, though its volatility has prompted regulatory scrutiny. Insecticides like DDT, featuring multiple chlorophenyl groups, exemplified early aryl halide applications in pest control before widespread bans due to bioaccumulation concerns.[95] Pentachlorophenol, a polychlorinated aryl compound, found use as a wood preservative and fungicide, leveraging its halogen content for antimicrobial effects. These examples underscore how aryl halides impart durability and biological activity, though modern developments favor fluorinated variants for improved environmental profiles.[96]Other Commercial Roles
Chlorobenzene functions as a versatile industrial solvent due to its high boiling point and chemical stability. It is commonly applied in degreasing operations for metal parts, formulation of paints and adhesives, and as a reaction medium in chemical manufacturing processes.[8] [97] Annual global production exceeds 10 million metric tons, with significant portions allocated to solvent applications despite regulatory scrutiny over volatility and toxicity.[98] Aryl halides also serve as intermediates in the synthesis of dyes and rubber processing agents. Chlorobenzene derivatives are incorporated into dyestuff production, enabling the creation of colorants for textiles and inks through subsequent substitution reactions.[98] In rubber manufacturing, chlorobenzene facilitates the production of vulcanization accelerators and other additives, enhancing material durability.[98] Certain brominated aryl compounds contribute to pigment formulations, where halogen substitution influences color stability and lightfastness. For example, dibromoindigo derivatives have been utilized in specialized pigments, though modern applications favor less persistent alternatives.[99]Environmental and Health Aspects
Toxicity Profiles and Exposure Risks
Aryl halides display variable toxicity profiles influenced by the halogen type, substitution pattern, and molecular structure, with simpler monohalides generally exhibiting lower acute potency compared to polyhalogenated congeners. Acute high-level exposures to compounds like chlorobenzene via inhalation or ingestion can induce central nervous system effects such as dizziness, headache, and drowsiness, alongside elevated liver enzymes indicative of hepatotoxicity and potential renal damage.[100] [101] Similarly, bromobenzene exposure irritates skin and mucous membranes, with systemic effects including liver and kidney impairment upon repeated or high-dose contact.[102] [11] These effects stem from metabolic activation to reactive intermediates that target hepatic and renal tissues, though aryl halides' relative stability limits reactivity relative to alkyl analogs.[100] Polyhalogenated aryl halides pose greater chronic risks due to persistence and bioaccumulation. Polychlorinated biphenyls (PCBs), consisting of biphenyl rings with multiple chlorine substitutions, are associated with liver cancer, malignant melanoma, thyroid dysfunction, and immunotoxicity in exposed workers and populations, with animal studies confirming carcinogenicity and reproductive toxicity.[103] [104] Dichlorodiphenyltrichloroethane (DDT), featuring chlorinated phenyl rings, demonstrates neurotoxic effects like tremors and seizures at high doses, alongside probable human carcinogenicity (particularly liver tumors) and links to breast cancer and decreased semen quality in epidemiological data.[105] [106] [107] Such congeners act as endocrine disruptors, mimicking hormones and altering metabolic pathways.[104] Occupational exposure risks predominate in chemical manufacturing and pesticide formulation, where inhalation of vapors or dermal contact during handling of aryl halides like chlorobenzene or bromobenzene can exceed safe thresholds, leading to the aforementioned acute effects; permissible exposure limits, such as 10 ppm for chlorobenzene (8-hour TWA), aim to mitigate these.[100] Environmental exposures for the general population arise primarily from legacy contamination, with PCBs bioaccumulating in fatty fish and meats, resulting in dietary intake levels prompting advisories (e.g., limited fish consumption near polluted sites).[103] DDT residues persist in soils and sediments, contributing to indirect exposure via food chains, though bans since the 1970s have reduced levels; vulnerable groups include children and pregnant women due to transgenerational risks like epigenetic changes.[107] Overall, while simple aryl halides present manageable industrial hazards with proper controls, persistent polyhalogenated variants drive long-term public health concerns through bioaccumulation and non-threshold carcinogenic mechanisms.[105][104]Persistence, Bioaccumulation, and Regulatory Responses
Polyhalogenated aryl halides, such as polychlorinated biphenyls (PCBs) and hexachlorobenzene (HCB), demonstrate significant environmental persistence owing to the stability of carbon-halogen bonds, which resist hydrolysis, photolysis, and microbial degradation under aerobic conditions.[108] These compounds can remain in soil and sediments for decades, with half-lives exceeding 10 years in many cases, as evidenced by their accumulation in anaerobic environments where reductive dehalogenation occurs slowly.[109] Simpler mono- or dihalogenated aryl compounds, like chlorobenzene, exhibit moderate persistence with half-lives in water ranging from days to months, but higher chlorination increases recalcitrance due to steric hindrance and electron-withdrawing effects that inhibit enzymatic attack.[110] Bioaccumulation of aryl halides is pronounced in lipophilic variants, which partition into fatty tissues and biomagnify through food chains, with bioconcentration factors often exceeding 10^4 in aquatic organisms for PCBs.[109] For instance, HCB shows a log Kow of approximately 5.5, facilitating uptake and retention in higher trophic levels, leading to elevated concentrations in predatory fish and mammals.[108] This potential is attributed to low water solubility and high octanol-water partition coefficients, though volatility limits accumulation in some gaseous phases.[109] Regulatory responses target persistent aryl halides classified as persistent organic pollutants (POPs) under the Stockholm Convention, which mandates elimination of production and use for PCBs, HCB, and pentachlorophenol, with specific exemptions for legacy equipment until 2025 or later under defined conditions.[111] The convention, effective since 2004 and ratified by over 180 parties, requires best available techniques for destruction of stockpiles and contaminated sites, driven by evidence of long-range transport and transboundary effects.[111] National implementations, such as U.S. EPA bans on PCB manufacturing since 1979 and EU restrictions under REACH, further enforce phase-outs and monitoring, prioritizing remediation of hotspots like transformer oils and pesticides. These measures reflect causal links between exposure and ecological disruption, overriding less stringent prior assessments influenced by industrial lobbying.[111]Biodegradation Challenges and Remediation Efforts
Aryl halides persist in the environment due to the thermodynamic stability of the carbon-halogen bond, particularly in aromatic systems where halogen substituents withdraw electrons and hinder nucleophilic attack or ring hydroxylation essential for microbial catabolism.[112] This recalcitrance limits aerobic degradation, as conventional dioxygenases struggle with electron-deficient substrates, often resulting in incomplete mineralization and accumulation of toxic partial degradation products.[112] For instance, polychlorinated biphenyls (PCBs) exhibit half-lives exceeding decades in sediments, with microbial transformation yielding lower-chlorinated congeners that bioaccumulate more readily despite reduced persistence.[113] Reductive dehalogenation under anaerobic conditions represents the primary microbial strategy for initiating aryl halide breakdown, wherein organohalide-respiring bacteria (OHRB) such as Dehalococcoides mccartyi employ cobalamin-dependent reductive dehalogenases (e.g., RdhA) to replace halogens with hydrogen, using the compounds as terminal electron acceptors.[112] This process demands specific electron donors like hydrogen or lactate and is constrained by narrow substrate specificity, competition among microbial consortia, and sensitivity to redox potentials, often yielding only partial dechlorination (e.g., hexa- to tri-chlorobiphenyls in PCBs).[114] Chlorobenzenes, similarly, undergo stepwise dehalogenation by strains like Desulfitobacterium spp., but rates remain slow without optimized conditions, exacerbating groundwater contamination.[115] Oxidative dehalogenation, while feasible for less halogenated aryl halides, faces enzymatic bottlenecks; monooxygenases (e.g., HadA in Ralstonia spp.) or dehalogenases (e.g., 4-chlorobenzoyl-CoA dehalogenase) enable halide replacement with hydroxyl groups, yet suffer from low turnover rates and inhibition by highly substituted rings.[112] Fluorinated aryl halides pose even greater barriers, as C-F bonds resist both reductive and oxidative assaults, underscoring the need for tailored microbial adaptations absent in most natural populations.[116] Remediation efforts emphasize sequential anaerobic-aerobic bioremediation, where OHRB first reduce halogen load (e.g., Dehalococcoides converting penta-chlorophenol to tri-chlorophenol), followed by aerobic strains like Pseudomonas putida for ring cleavage via biphenyl dioxygenases.[114] Bioaugmentation with enriched consortia has accelerated PCB dechlorination in lab mesocosms by up to 10-fold when supplemented with electron donors, though field-scale applications contend with bioavailability issues and incomplete pathways.[113] Genetic engineering of dehalogenases, such as introducing stabilizing mutations (e.g., G513Y in PcpB), enhances efficiency for pollutants like pentachlorophenol, while hybrid physico-biological approaches (e.g., zero-valent iron priming for dehalogenation) address limitations in enzyme stability and substrate range.[112]Recent Advances
Innovations in Selective Functionalization
Innovations in selective functionalization of aryl halides have primarily targeted regioselective cross-couplings in polyhalogenated substrates, where identical halogens complicate traditional methods by yielding statistical mixtures. Strategies leverage electronic effects, steric hindrance, directing groups, and ligand modifications to dictate site specificity, often achieving high selectivity (>90% in many cases) without protecting groups. A comprehensive review highlights these approaches, including catalyst tuning for ortho or meta preference in dihalobenzenes and phenols.[117] Ligand-controlled methods exemplify progress; for instance, sulfonylated phosphine ligands like sSPhos or sXPhos promote meta-selective Suzuki-Miyaura couplings via electrostatic guidance from proximal ammonium or carbonyl groups, yielding up to 7.5:1 meta:ortho ratios in reactions of N-acetyl-3,4-dichlorobenzylamine derivatives with arylboronic acids under Pd catalysis in 2016. Similarly, bifunctional HTP/DHTP ligands enable ortho-selective Kumada or Sonogashira couplings in 2,4-dibromophenols (>90% ortho selectivity) by coordinating the phenolic OH to direct Pd, as reported between 2009 and 2014. Directing groups, such as amines in 2-amino-3,5-dibromopyrazines, guide Stille couplings to specific sites via coordination, achieving clean C3 selectivity since early examples in 1995, with ongoing refinements. Alternative innovations circumvent direct selectivity through halide migration; base-catalyzed isomerization of 3-bromopyridines to 4-bromopyridines via pyridyne intermediates, using P4-t-Bu or KOH/18-crown-6 in dioxane or DMA, enables subsequent 4-selective SNAr etherification, hydroxylation, or amination with alcohols or amines, attaining >14:1 regioselectivity and yields up to 76% in 2020.[118] These methods extend to aryl systems, reducing over-functionalization risks in complex syntheses. Emerging dual-catalysis approaches, such as Ni/photoredox systems, further enhance site control in couplings with bis-boronic esters, though detailed scopes emphasize heteroaryl extensions.[119] Overall, these advances prioritize catalyst efficiency over substrate premodification, aligning with sustainable synthesis goals.Sustainable and Green Chemistry Developments
Efforts in green chemistry for aryl halides focus on replacing energy-intensive, solvent-heavy processes with bio-based feedstocks, earth-abundant catalysts, and solvent-minimized techniques to reduce environmental footprints. A key advancement involves deriving aryl halides from lignin, an abundant lignocellulosic biomass byproduct, through selective depolymerization and halogenation under mild conditions, yielding up to 40% isolated products from technical lignins without petroleum reliance.[120] This approach enhances sustainability by valorizing waste streams, with mechanistic studies confirming radical-mediated C-H halogenation as the core step.[120] Cross-coupling reactions, central to aryl halide functionalization, have seen eco-friendly innovations such as copper-free Heck–Cassar–Sonogashira and Suzuki–Miyaura variants conducted in aqueous or solvent-free media, achieving yields over 90% for diverse substrates while avoiding toxic ligands and precious metals.[121] Electrochemical protocols further promote sustainability by enabling C-C and C-N couplings of aryl halides using electricity as the reductant, eliminating sacrificial reagents and operating in water at room temperature with turnover numbers exceeding 1000.[122] These methods prioritize atom economy, with recent examples incorporating air-stable nickel catalysts for broad substrate tolerance.[122] Carbonylative transformations of aryl halides using first-row transition metals like iron or cobalt provide greener routes to acyl compounds, proceeding in biomass-derived solvents with minimal waste and catalyst loadings below 1 mol%.[123] Mechanochemical ball-milling enables solvent-free radical reductions of aryl halides, converting them to arenes in minutes with quantitative yields and recyclability, circumventing volatile organic compounds. Metal-free alternatives, such as magnetized water-promoted N-arylations, further align with green principles by using benign media and avoiding transition metals altogether.[124]References
- https://pubmed.ncbi.nlm.nih.gov/33533358/