
Community hub
Recent from talks
Contribute something
Nothing was collected or created yet.
Papillomaviridae
View on Wikipedia| Papillomaviridae | |
|---|---|

| |
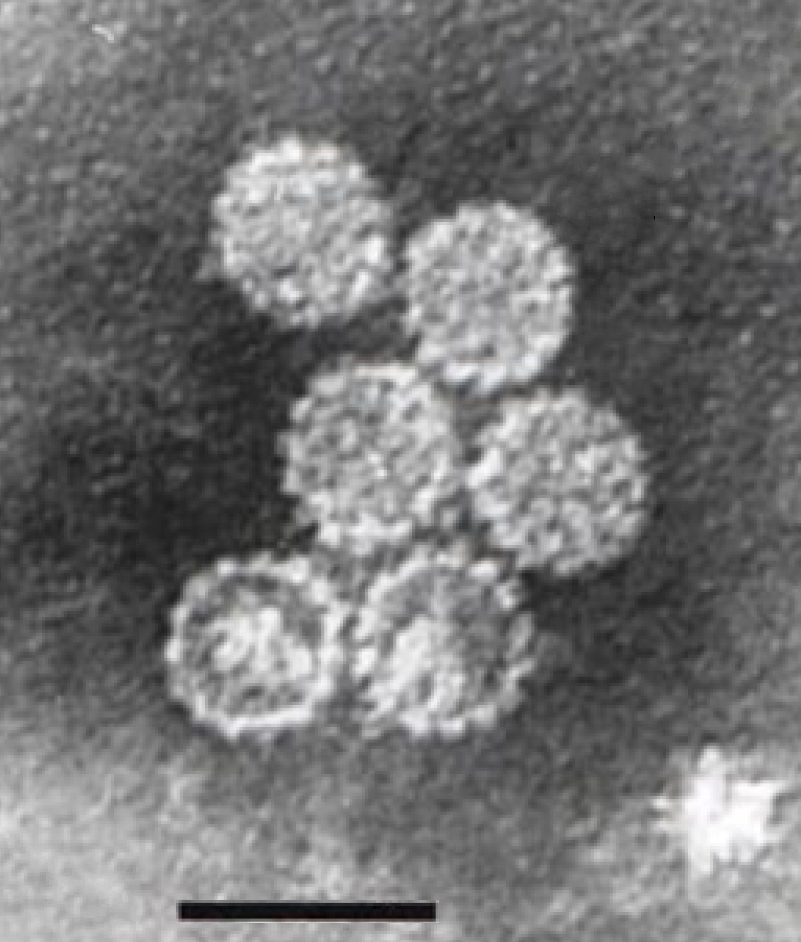
| Electron micrograph of papillomavirus, scale bar 70 nm | |
| Virus classification | |
| (unranked): | Virus |
| Realm: | Monodnaviria |
| Kingdom: | Shotokuvirae |
| Phylum: | Cossaviricota |
| Class: | Papovaviricetes |
| Order: | Zurhausenvirales |
| Family: | Papillomaviridae |
| Subfamilies and genera | |
Papillomaviridae is a family of non-enveloped double-stranded DNA viruses whose members are known as papillomaviruses.[1] Several hundred species of papillomaviruses, traditionally referred to as "types",[2] have been identified infecting all carefully inspected mammals,[2] but also other vertebrates such as birds, snakes, turtles and fish.[3][4][5] Infection by most papillomavirus types, depending on the type, is either asymptomatic (e.g. most Beta-PVs) or causes small benign tumors, known as papillomas or warts (e.g. human papillomavirus 1, HPV6 or HPV11). Papillomas caused by some types, however, such as human papillomaviruses 16 and 18, carry a risk of becoming cancerous.[6]
Papillomaviruses are usually considered as highly host- and tissue-tropic, and are thought to rarely be transmitted between species.[7] Papillomaviruses replicate exclusively in the basal layer of the body surface tissues. All known papillomavirus types infect a particular body surface,[2] typically the skin or mucosal epithelium of the genitals, anus, mouth, or airways.[8] For example, human papillomavirus (HPV) type 1 tends to infect the soles of the feet, and HPV type 2 the palms of the hands, where they may cause warts. Additionally, there are descriptions of the presence of papillomavirus DNA in the blood and in the peripheral blood mononuclear cells.
Papillomaviruses were first identified in the early 20th century, when it was shown that skin warts, or papillomas, could be transmitted between individuals by a filterable infectious agent. In 1935 Francis Peyton Rous, who had previously demonstrated the existence of a cancer-causing sarcoma virus in chickens, went on to show that a papillomavirus could cause skin cancer in infected rabbits. This was the first demonstration that a virus could cause cancer in mammals.
Taxonomy of papillomaviruses
[edit]
There are over 100 species of papillomavirus recognised,[9] though the ICTV officially recognizes a smaller number, categorized into 53 genera, as of 2019.[10][11][12] All papillomaviruses (PVs) have similar genomic organizations, and any pair of PVs contains at least five homologous genes, although the nucleotide sequence may diverge by more than 50%. Phylogenetic algorithms that permit the comparison of homologies led to phylogenetic trees that have a similar topology, independent of the gene analyzed.[13]
Phylogenetic studies strongly suggest that PVs normally evolve together with their mammalian and bird host species, but adaptive radiations, occasional zoonotic events and recombinations may also impact their diversification.[13] Their basic genomic organization appears maintained for a period exceeding 100 million years, and these sequence comparisons have laid the foundation for a PV taxonomy, which is now officially recognized by the International Committee on Taxonomy of Viruses. All PVs form the family Papillomaviridae, which is distinct from the Polyomaviridae thus eliminating the term Papovaviridae. Major branches of the phylogenetic tree of PVs are considered genera, which are identified by Greek letters. Minor branches are considered species and unite PV types that are genomically distinct without exhibiting known biological differences. This new taxonomic system does not affect the traditional identification and characterization of PV "types" and their independent isolates with minor genomic differences, referred to as "subtypes" and "variants", all of which are taxa below the level of "species".[14] Additionally, phylogenetic groupings at higher taxonomic level have been proposed.[15]
This classification may need revision in the light of the existence of papilloma–polyoma virus recombinants.[16] Additional species have also been described. Sparus aurata papillomavirus 1 has been isolated from fish.[17]
The family contains two subfamilies and 53 genera, listed hereafter (-virinae denotes subfamilies and -virus denotes genera):[18]
Subfamily: Firstpapillomavirinae
- Alphapapillomavirus
- Betapapillomavirus
- Chipapillomavirus
- Deltapapillomavirus
- Dyochipapillomavirus
- Dyodeltapapillomavirus
- Dyoepsilonpapillomavirus
- Dyoetapapillomavirus
- Dyoiotapapillomavirus
- Dyokappapapillomavirus
- Dyolambdapapillomavirus
- Dyomupapillomavirus
- Dyonupapillomavirus
- Dyoomegapapillomavirus
- Dyoomikronpapillomavirus
- Dyophipapillomavirus
- Dyopipapillomavirus
- Dyopsipapillomavirus
- Dyorhopapillomavirus
- Dyosigmapapillomavirus
- Dyotaupapillomavirus
- Dyothetapapillomavirus
- Dyoupsilonpapillomavirus
- Dyoxipapillomavirus
- Dyozetapapillomavirus
- Epsilonpapillomavirus
- Etapapillomavirus
- Gammapapillomavirus
- Iotapapillomavirus
- Kappapapillomavirus
- Lambdapapillomavirus
- Mupapillomavirus
- Nupapillomavirus
- Omegapapillomavirus
- Omikronpapillomavirus
- Phipapillomavirus
- Pipapillomavirus
- Psipapillomavirus
- Rhopapillomavirus
- Sigmapapillomavirus
- Taupapillomavirus
- Thetapapillomavirus
- Treisdeltapapillomavirus
- Treisepsilonpapillomavirus
- Treisetapapillomavirus
- Treisiotapapillomavirus
- Treiskappapapillomavirus
- Treisthetapapillomavirus
- Treiszetapapillomavirus
- Upsilonpapillomavirus
- Xipapillomavirus
- Zetapapillomavirus
- Subfamily: Secondpapillomavirinae
Human papillomaviruses
[edit]In 2014, 174 human papillomavirus types had been completely sequenced according to the International Human Papillomavirus Reference Center.[19][20] They have been divided into five genera: Alphapapillomavirus, Betapapillomavirus, Gammapapillomavirus, Mupapillomavirus and Nupapillomavirus.[19] At least 200 additional viruses have been identified that await sequencing and classification.[citation needed]
Animal papillomaviruses
[edit]
Around 280 papillomaviruses have been identified in other animals, with mammalian, avian, reptilian, and fish hosts.[21][22]
Individual papillomavirus types tend to be highly adapted to replication in a single animal species. In one study, researchers swabbed the forehead skin of a variety of zoo animals and used PCR to amplify any papillomavirus DNA that might be present.[23] Although a wide variety of papillomavirus sequences were identified in the study, the authors found little evidence for inter-species transmission. One zookeeper was found to be transiently positive for a chimpanzee-specific papillomavirus sequence. However, the authors note that the chimpanzee-specific papillomavirus sequence could have been the result of surface contamination of the zookeeper's skin, as opposed to productive infection.[citation needed]
Cottontail rabbit papillomavirus (CRPV) can cause protuberant warts in its native host, the North American rabbit genus Sylvilagus. These horn-like warts may be the original basis for the urban legends of the American antlered rabbit the Jackalope and European Wolpertinger.[24] European domestic rabbits (genus Oryctolagus) can be transiently infected with CRPV in a laboratory setting. However, since European domestic rabbits do not produce infectious progeny virus, they are considered an incidental or "dead-end" host for CRPV.[25]
Inter-species transmission has also been documented for bovine papillomavirus (BPV) type 1.[26] In its natural host (cattle), BPV-1 induces large fibrous skin warts. BPV-1 infection of horses, which are an incidental host for the virus, can lead to the development of benign tumors known as sarcoids. The agricultural significance of BPV-1 spurred a successful effort to develop a vaccine against the virus.[citation needed]
A few reports have identified papillomaviruses in smaller rodents, such as Syrian hamsters, the African multimammate rat and the Eurasian harvest mouse.[27] However, there are no papillomaviruses known to be capable of infecting laboratory mice. The lack of a tractable mouse model for papillomavirus infection has been a major limitation for laboratory investigation of papillomaviruses.[citation needed]
Twenty types have been identified in seals.[22]
Four papillomaviruses are known to infect birds: Fringilla coelebs papillomavirus 1, Francolinus leucoscepus papillomavirus 1, Psittacus erithacus papillomavirus 1 and Pygoscelis adeliae papillomavirus 1.[28] All these species have a gene (E9) of unknown function, suggesting a common origin.
Evolution
[edit]The evolution of papillomaviruses is thought to be slow compared to many other virus types, but there are no experimental measurements currently available. This is probably because the papillomavirus genome is composed of genetically stable double-stranded DNA that is replicated with high fidelity by the host cell's DNA replication machinery.[citation needed]
It is believed that papillomaviruses generally co-evolve with a particular species of host animal over many years, although there are strong evidences against the hypothesis of coevolution.[13][29] In a particularly speedy example, HPV-16 has evolved slightly as human populations have expanded across the globe and now varies in different geographic regions in a way that probably reflects the history of human migration.[30][31] Cutaneotropic HPV types are occasionally exchanged between family members during the entire lifetime, but other donors should also be considered in viral transmission.[32]
Other HPV types, such as HPV-13, vary relatively little in different human populations. In fact, the sequence of HPV-13 closely resembles a papillomavirus of bonobos (also known as pygmy chimpanzees).[33] It is not clear whether this similarity is due to recent transmission between species or because HPV-13 has simply changed very little in the six or so million years since humans and bonobos diverged.[31]
The most recent common ancestor of this group of viruses has been estimated to have existed 424 million years ago.[34]
There are five main genera infecting humans (Alpha, Beta, Gamma, Mu and Nu). The most recent common ancestor of these genera evolved 49.7 million years ago-58.5 million years ago.[35] The most recent ancestor of the gamma genus was estimated to have evolved between 45.3 million years ago and 67.5 million years ago.[citation needed]
Structure
[edit]
Papillomaviruses are non-enveloped, meaning that the outer shell or capsid of the virus is not covered by a lipid membrane. A single viral protein, known as L1, is necessary and sufficient for formation of a 55–60 nanometer capsid composed of 72 star-shaped capsomers (see figure). Like most non-enveloped viruses, the capsid is geometrically regular and presents icosahedral symmetry. Self-assembled virus-like particles composed of L1 are the basis of a successful group of prophylactic HPV vaccines designed to elicit virus-neutralizing antibodies that protect against initial HPV infection. As such, papillomaviridæ are stable in the environment.[citation needed]
The papillomavirus genome is a double-stranded circular DNA molecule ~8,000 base pairs in length. It is packaged within the L1 shell along with cellular histone proteins, which serve to wrap and condense DNA.[citation needed]
The papillomavirus capsid also contains a viral protein known as L2, which is less abundant. Although not clear how L2 is arranged within the virion, it is known to perform several important functions, including facilitating the packaging of the viral genome into nascent virions as well as the infectious entry of the virus into new host cells. L2 is of interest as a possible target for more broadly protective HPV vaccines.
The viral capsid consists of 72 capsomeres of which 12 are five-coordinated and 60 are six-coordinated capsomeres, arranged on a T = 7d icosahedral surface lattice.[36]
Tissue specificity
[edit]Papillomaviruses replicate exclusively in keratinocytes. Keratinocytes form the outermost layers of the skin, as well as some mucosal surfaces, such as the inside of the cheek or the walls of the vagina. These surface tissues, which are known as stratified squamous epithelia, are composed of stacked layers of flattened cells. The cell layers are formed through a process known as cellular differentiation, in which keratinocytes gradually become specialized, eventually forming a hard, crosslinked surface that prevents moisture loss and acts as a barrier against pathogens. Less-differentiated keratinocyte stem cells, replenished on the surface layer, are thought to be the initial target of productive papillomavirus infections. Subsequent steps in the viral life cycle are strictly dependent on the process of keratinocyte differentiation. As a result, papillomaviruses can only replicate in body surface tissues.[citation needed]
Life cycle
[edit]Infectious entry
[edit]Papillomaviruses gain access to keratinocyte stem cells through small wounds, known as microtraumas, in the skin or mucosal surface. Interactions between L1 and sulfated sugars on the cell surface promote initial attachment of the virus.[37][38] The virus is then able to get inside from the cell surface via interaction with a specific receptor, likely via the alpha-6 beta-4 integrin,[39][40] and transported to membrane-enclosed vesicles called endosomes.[41][42] The capsid protein L2 disrupts the membrane of the endosome through a cationic cell-penetrating peptide, allowing the viral genome to escape and traffic, along with L2, to the cell nucleus.[43][44][45]
Viral persistence and latency
[edit]After successful infection of a keratinocyte, the virus expresses E1 and E2 proteins, which are for replicating and maintaining the viral DNA as a circular episome. The viral oncogenes E6 and E7 promote cell growth by inactivating the tumor suppressor proteins p53 and pRb. Keratinocyte stem cells in the epithelial basement layer can maintain papillomavirus genomes for decades.[8]
Production of progeny virus
[edit]The current understanding is that viral DNA replication likely occurs in the G2 phase of the cell cycle and rely on recombination-dependent replication supported by DNA damage response mechanisms (activated by the E7 protein) to produce progeny viral genomes.[46] Papillomavirus genomes are sometimes integrated into the host genome, especially noticeable with oncogenic HPVs, but is not a normal part of the virus life cycle and a dead-end that eliminates the potential of viral progeny production.[46]
The expression of the viral late genes, L1 and L2, is exclusively restricted to differentiating keratinocytes in the outermost layers of the skin or mucosal surface. The increased expression of L1 and L2 is typically correlated with a dramatic increase in the number of copies of the viral genome. Since the outer layers of stratified squamous epithelia are subject to relatively limited surveillance by cells of the immune system, it is thought that this restriction of viral late gene expression represents a form of immune evasion.[citation needed]
New infectious progeny viruses are assembled in the cell nucleus. Papillomaviruses have evolved a mechanism for releasing virions into the environment. Other kinds of non-enveloped animal viruses utilize an active lytic process to kill the host cell, allowing release of progeny virus particles. Often this lytic process is associated with inflammation, which might trigger immune attack against the virus. Papillomaviruses exploit desquamation as a stealthy, non-inflammatory release mechanism.[citation needed]
| Genus | Host details | Tissue tropism | Entry details | Release details | Replication site | Assembly site | Transmission |
|---|---|---|---|---|---|---|---|
| Dyoxipapillomavirus | Vertebrates | None | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Omikronpapillomavirus | Porpoises | Epithelial: mucous; epithelial: skin | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Dyodeltapapillomavirus | Vertebrates | None | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Omegapapillomavirus | Vertebrates | Epithelial: mucous; epithelial: skin | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Nupapillomavirus | Humans | Epithelial: mucous; epithelial: skin | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Dyomupapillomavirus | Vertebrates | None | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Dyozetapapillomavirus | Vertebrates | None | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Kappapapillomavirus | Rabbits | Epithelial: mucous; epithelial: skin | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Upsilonpapillomavirus | Vertebrates | Epithelial: mucous; epithelial: skin | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Dyoetapapillomavirus | Vertebrates | None | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Sigmapapillomavirus | Vertebrates | Epithelial: mucous; epithelial: skin | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Lambdapapillomavirus | Cats; dogs | Epithelial: mucous; epithelial: skin | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Taupapillomavirus | Vertebrates | Epithelial: mucous; epithelial: skin | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Betapapillomavirus | Humans | Epithelial: mucous; epithelial: skin | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Xipapillomavirus | Bovines | Epithelial: mucous; epithelial: skin | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Dyoepsilonpapillomavirus | Vertebrates | None | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Thetapapillomavirus | Birds | Epithelial: mucous; epithelial: skin | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Etapapillomavirus | Birds | Epithelial: skin | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Rhopapillomavirus | Vertebrates | Epithelial: mucous; epithelial: skin | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Dyothetapapillomavirus | Vertebrates | None | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Dyoomikronpapillomavirus | Vertebrates | None | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Gammapapillomavirus | Humans | Epithelial: mucous; epithelial: skin | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Alphapapillomavirus | Humans; monkeys | Epithelial: mucous; epithelial: skin | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Sex; contact |
| Zetapapillomavirus | Horses | Epithelial: mucous; epithelial: skin | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Deltapapillomavirus | Ruminants | Epithelial: mucous; epithelial: skin | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Dyolambdapapillomavirus | Vertebrates | None | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Dyosigmapapillomavirus | Vertebrates | None | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Dyorhopapillomavirus | Vertebrates | None | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Psipapillomavirus | Vertebrates | Epithelial: mucous; epithelial: skin | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Dyokappapapillomavirus | Vertebrates | None | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Pipapillomavirus | Hamsters | Epithelial: mucous; epithelial: skin | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Iotapapillomavirus | Rodents | Epithelial: mucous; epithelial: skin | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Epsilonpapillomavirus | Bovines | Epithelial: skin | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Phipapillomavirus | Vertebrates | Epithelial: mucous; epithelial: skin | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Dyonupapillomavirus | Vertebrates | None | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Dyopipapillomavirus | Vertebrates | None | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Dyoiotapapillomavirus | Vertebrates | None | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
| Mupapillomavirus | Humans | Epithelial: mucous; epithelial: skin | Cell receptor endocytosis | Lysis | Nucleus | Nucleus | Contact |
Association with cancer
[edit]Although some papillomavirus types can cause cancer in the epithelial tissues they inhabit, cancer is not a typical outcome of infection. The development of papillomavirus-induced cancers typically occurs over the course of many years. Papillomaviruses have been associated with the development of cervical cancer, penile cancer[47] and oral cancers.[48] An association with vulval cancer and urothelial carcinoma with squamous differentiation in patients with neurogenic bladder has also been noted.[49][50] There are cancer causing papillomavirus genome that encodes two small proteins called E6 and E7 that mimic cancer causing oncogenes. The way they work is that they stimulate unnatural growth of cells and block their natural defenses. Also they act on many signaling proteins that control proliferation and apoptosis.[51]
Laboratory study
[edit]The fact that the papillomavirus life cycle strictly requires keratinocyte differentiation has posed a substantial barrier to the study of papillomaviruses in the laboratory, since it has precluded the use of conventional cell lines to grow the viruses. Because infectious BPV-1 virions can be extracted from the large warts the virus induces on cattle, it has been a workhorse model papillomavirus type for many years. CRPV, rabbit oral papillomavirus (ROPV) and canine oral papillomavirus (COPV) have also been used extensively for laboratory studies. As soon as researchers discovered that these viruses cause cancer, they worked together to find a vaccine to it. Currently, the most effective way to go about it is to mimic a virus that is composed of L1 protein but lack the DNA. Basically, our immune system builds defenses against infections, but if these infections do not cause disease they can be used as a vaccine. PDB entry 6bt3 shows how antibodies surfaces attack the surface of the virus to disable it.[52]
Some sexually transmitted HPV types have been propagated using a mouse "xenograft" system, in which HPV-infected human cells are implanted into immunodeficient mice. More recently, some groups have succeeded in isolating infectious HPV-16 from human cervical lesions. However, isolation of infectious virions using this technique is arduous and the yield of infectious virus is very low.[citation needed]
The differentiation of keratinocytes can be mimicked in vitro by exposing cultured keratinocytes to an air/liquid interface. The adaptation of such "raft culture" systems to the study of papillomaviruses was a significant breakthrough for in vitro study of the viral life cycle.[53] However, raft culture systems are relatively cumbersome and the yield of infectious HPVs can be low.[54]
The development of a yeast-based system that allows stable episomal HPV replication provides a convenient, rapid and inexpensive means to study several aspects of the HPV lifecycle (Angeletti 2002). For example, E2-dependent transcription, genome amplification and efficient encapsidation of full-length HPV DNAs can be easily recreated in yeast (Angeletti 2005).
Recently, transient high-yield methods for producing HPV pseudoviruses carrying reporter genes has been developed. Although pseudoviruses are not suitable for studying certain aspects of the viral life cycle, initial studies suggest that their structure and initial infectious entry into cells is probably similar in many ways to authentic papillomaviruses.
Human papillomavirus binds to heparin molecules on the surface of the cells that it infects. Studies have shown that the crystal of isolated L1 capsomeres has the heparin chains recognized by lysines lines grooves on the surface of the virus. Also those with the antibodies show that they can block this recognition.[55]
Genetic organization and gene expression
[edit]
The papillomavirus genome is divided into an early region (E), encoding six open reading frames (ORF) (E1, E2, E4, E5, E6, and E7) that are expressed immediately after initial infection of a host cell, and a late region (L) encoding a major capsid protein L1 and a minor capsid protein L2. All viral ORFs are encoded on one DNA strand (see figure). This represents a dramatic difference between papillomaviruses and polyomaviruses, since the latter virus type expresses its early and late genes by bi-directional transcription of both DNA strands. This difference was a major factor in establishment of the consensus that papillomaviruses and polyomaviruses probably never shared a common ancestor, despite the striking similarities in the structures of their virions.[citation needed]
After the host cell is infected, HPV16 early promoter is activated and a polycistronic primary RNA containing all six early ORFs is transcribed. This polycistronic RNA contains three exons and two introns and undergoes active RNA splicing to generate multiple isoforms of mRNAs.[56] One of the spliced isoform RNAs, E6*I, serves as an E7 mRNA to translate E7 oncoprotein.[57] In contrast, an intron in the E6 ORF that remains intact without splicing is necessary for translation of E6 oncoprotein.[57] However, viral early transcription subjects to viral E2 regulation and high E2 levels repress the transcription. HPV genomes integrate into host genome by disruption of E2 ORF, preventing E2 repression on E6 and E7. Thus, viral genome integration into host DNA genome increases E6 and E7 expression to promote cellular proliferation and the chance of malignancy.[citation needed]
A major viral late promoter in viral early region becomes active only in differentiated cells and its activity can be highly enhanced by viral DNA replication. The late transcript is also a polycistronic RNA which contains two introns and three exons. Alternative RNA Splicing of this late transcript is essential for L1 and L2 expression and can be regulated by RNA cis-elements and host splicing factors.[56][58][59]
Technical discussion of papillomavirus gene functions
[edit]Genes within the papillomavirus genome are usually identified after similarity with other previously identified genes. However, some spurious open reading frames might have been mistaken as genes simply after their position in the genome, and might not be true genes. This applies specially to certain E3, E4, E5 and E8 open reading frames.[citation needed]
E1
[edit]Encodes a protein that binds to the viral origin of replication in the long control region of the viral genome. E1 uses ATP to exert a helicase activity that forces apart the DNA strands, thus preparing the viral genome for replication by cellular DNA replication factors.
E2
[edit]The E2 protein serves as a master transcriptional regulator for viral promoters located primarily in the long control region. The protein has a transactivation domain linked by a relatively unstructured hinge region to a well-characterized DNA binding domain. E2 facilitates the binding of E1 to the viral origin of replication. E2 also utilizes a cellular protein known as Bromodomain-4 (Brd4) to tether the viral genome to cellular chromosomes.[60] This tethering to the cell's nuclear matrix ensures faithful distribution of viral genomes to each daughter cell after cell division. It is thought that E2 serves as a negative regulator of expression for the oncogenes E6 and E7 in latently HPV-infected basal layer keratinocytes. Genetic changes, such as integration of the viral DNA into a host cell chromosome, that inactivate E2 expression tend to increase the expression of the E6 and E7 oncogenes, resulting in cellular transformation and possibly further genetic destabilization.
E3
[edit]This small putative gene exists only in a few papillomavirus types. The gene is not known to be expressed as a protein and does not appear to serve any function.
E4
[edit]Although E4 proteins are expressed at low levels during the early phase of viral infection, expression of E4 increases dramatically during the late phase of infection. In other words, its "E" appellation may be something of a misnomer. In the case of HPV-1, E4 can account for up to 30% of the total protein at the surface of a wart.[61] The E4 protein of many papillomavirus types is thought to facilitate virion release into the environment by disrupting intermediate filaments of the keratinocyte cytoskeleton. Viral mutants incapable of expressing E4 do not support high-level replication of the viral DNA, but it is not yet clear how E4 facilitates DNA replication. E4 has also been shown to participate in arresting cells in the G2 phase of the cell cycle.
E5
[edit]The E5 are small, very hydrophobic proteins that destabilise the function of many membrane proteins in the infected cell.[62] The E5 protein of some animal papillomavirus types (mainly bovine papillomavirus type 1) functions as an oncogene primarily by activating the cell growth-promoting signaling of platelet-derived growth factor receptors. The E5 proteins of human papillomaviruses associated to cancer, however, seem to activate the signal cascade initiated by epidermal growth factor upon ligand binding. HPV16 E5 and HPV2 E5 have also been shown to down-regulate the surface expression of major histocompatibility complex class I proteins, which may prevent the infected cell from being eliminated by killer T cells.
E6
[edit]
E6 is a 151 amino-acid peptide that incorporates a type 1 motif with a consensus sequence –(T/S)-(X)-(V/I)-COOH.[64][65] It also has two zinc finger motifs.[64]
E6 is of particular interest because it appears to have multiple roles in the cell and to interact with many other proteins. Its major role, however, is to mediate the degradation of p53, a major tumor suppressor protein, reducing the cell's ability to respond to DNA damage.[66][67]
E6 has also been shown to target other cellular proteins, thereby altering several metabolic pathways. One such target is NFX1-91, which normally represses production of telomerase, a protein that allows cells to divide an unlimited number of times. When NFX1-91 is degraded by E6, telomerase levels increase, inactivating a major mechanism keeping cell growth in check.[68] Additionally, E6 can act as a transcriptional cofactor—specifically, a transcription activator—when interacting with the cellular transcription factor, E2F1/DP1.[64]
E6 can also bind to PDZ-domains, short sequences which are often found in signaling proteins. E6's structural motif allows for interaction with PDZ domains on DLG (discs large) and hDLG (Drosophila large) tumor suppressor genes.[65][69] Binding at these locations causes transformation of the DLG protein and disruption of its suppressor function. E6 proteins also interact with the MAGUK (membrane-associated guanylate kinase family) proteins. These proteins, including MAGI-1, MAGI-2, and MAGI-3 are usually structural proteins, and can help with signaling.[65][69] More significantly, they are believed to be involved with DLG's suppression activity. When E6 complexes with the PDZ domains on the MAGI proteins, it distorts their shape and thereby impedes their function. Overall, the E6 protein serves to impede normal protein activity in such a way as to allow a cell to grow and multiply at the increased rate characteristic of cancer.
Since the expression of E6 is strictly required for maintenance of a malignant phenotype in HPV-induced cancers, it is an appealing target of therapeutic HPV vaccines designed to eradicate established cervical cancer tumors.
E7
[edit]In most papillomavirus types, the primary function of the E7 protein is to inactivate members of the pRb family of tumor suppressor proteins. Together with E6, E7 serves to prevent cell death (apoptosis) and promote cell cycle progression, thus priming the cell for replication of the viral DNA. E7 also participates in immortalization of infected cells by activating cellular telomerase. Like E6, E7 is the subject of intense research interest and is believed to exert a wide variety of other effects on infected cells. As with E6, the ongoing expression of E7 is required for survival of cancer cell lines, such as HeLa, that are derived from HPV-induced tumors.[70]
E8
[edit]Only a few papillomavirus types encode a short protein from the E8 gene. In the case of BPV-4 (papillomavirus genus Xi), the E8 open reading frame may substitute for the E6 open reading frame, which is absent in this papillomavirus genus.[71] These E8 genes are chemically and functionally similar to the E5 genes from some human papillomaviruses, and are also called E5/E8.
L1
[edit]L1 spontaneously self-assembles into pentameric capsomers. Purified capsomers can go on to form capsids, which are stabilized by disulfide bonds between neighboring L1 molecules. L1 capsids assembled in vitro are the basis of prophylactic vaccines against several HPV types. Compared to other papillomavirus genes, the amino acid sequences of most portions of L1 are well-conserved between types. However, the surface loops of L1 can differ substantially, even for different members of a particular papillomavirus species. This probably reflects a mechanism for evasion of neutralizing antibody responses elicited by previous papillomavirus infections.[72]
L2
[edit]L2 exists in an oxidized state within the papillomavirus virion, with the two conserved cysteine residues forming an intramolecular disulfide bond.[73] In addition to cooperating with L1 to package the viral DNA into the virion, L2 has been shown to interact with a number of cellular proteins during the infectious entry process. After the initial binding of the virion to the cell, L2 must be cleaved by the cellular protease furin.[74] The virion is internalized, probably through a clathrin-mediated process, into an endosome, where acidic conditions are thought to lead to exposure of membrane-destabilizing portions of L2.[43] The cellular proteins beta-actin[75] and syntaxin-18[76] may also participate in L2-mediated entry events. After endosome escape, L2 and the viral genome are imported into the cell nucleus where they traffic to a sub-nuclear domain known as an ND-10 body that is rich in transcription factors.[44] Small portions of L2 are well-conserved between different papillomavirus types, and experimental vaccines targeting these conserved domains may offer protection against a broad range of HPV types.[77]
See also
[edit]References
[edit]- ^ Van Doorslaer, K; Chen, Z; Bernard, HU; Chan, PKS; DeSalle, R; Dillner, J; Forslund, O; Haga, T; McBride, AA; Villa, LL; Burk, RD; Ictv Report, Consortium (August 2018). "ICTV Virus Taxonomy Profile: Papillomaviridae". The Journal of General Virology. 99 (8): 989–990. doi:10.1099/jgv.0.001105. PMC 6171710. PMID 29927370.
- ^ a b c de Villiers EM, Fauquet C, Broker TR, Bernard HU, zur Hausen H (June 2004). "Classification of papillomaviruses". Virology. 324 (1): 17–27. doi:10.1016/j.virol.2004.03.033. PMID 15183049.
- ^ Herbst LH, Lenz J, Van Doorslaer K, Chen Z, Stacy BA, Wellehan JF, Manire CA, Burk RD (January 2009). "Genomic characterization of two novel reptilian papillomaviruses, Chelonia mydas papillomavirus 1 and Caretta caretta papillomavirus 1". Virology. 383 (1): 131–5. doi:10.1016/j.virol.2008.09.022. PMID 18973915.
- ^ Drury SE, Gough RE, McArthur S, Jessop M (December 1998). "Detection of herpesvirus-like and papillomavirus-like particles associated with diseases of tortoises". The Veterinary Record. 143 (23): 639. PMID 9881444.
- ^ Lange CE, Favrot C, Ackermann M, Gull J, Vetsch E, Tobler K (September 2011). "Novel snake papillomavirus does not cluster with other non-mammalian papillomaviruses". Virology Journal. 8: 436. doi:10.1186/1743-422X-8-436. PMC 3179961. PMID 21910860.
- ^ Muñoz N, Castellsagué X, de González AB, Gissmann L (August 2006). "Chapter 1: HPV in the etiology of human cancer". Vaccine. 24 Suppl 3 (3): S3/1–10. doi:10.1016/j.vaccine.2006.05.115. PMID 16949995.
- ^ Mistry N, Wibom C, Evander M (October 2008). "Cutaneous and mucosal human papillomaviruses differ in net surface charge, potential impact on tropism". Virology Journal. 5: 118. doi:10.1186/1743-422X-5-118. PMC 2571092. PMID 18854037.
- ^ a b Doorbar J (March 2005). "The papillomavirus life cycle". Journal of Clinical Virology. 32 (Suppl 1): S7–15. doi:10.1016/j.jcv.2004.12.006. PMID 15753007.
- ^ Kocjan BJ, Hosnjak L, Seme K, Poljak M (May 2013). "Complete Genome Sequence of a Novel Human Betapapillomavirus, HPV-159". Genome Announcements. 1 (3) e00298–13. doi:10.1128/genomeA.00298-13. PMC 3668007. PMID 23723399.
- ^ "Virus Taxonomy: 2018b Release". International Committee on Taxonomy of Viruses. February 2019. Retrieved 26 March 2019.
- ^ "Virus Taxonomy: 2014 Release". ICTV. Retrieved 15 June 2015.
- ^ Bernard HU, Burk RD, Chen Z, van Doorslaer K, zur Hausen H, de Villiers EM (May 2010). "Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments". Virology. 401 (1): 70–9. doi:10.1016/j.virol.2010.02.002. PMC 3400342. PMID 20206957.
- ^ a b c Gottschling M, Stamatakis A, Nindl I, Stockfleth E, Alonso Á, Bravo IG (2007). "Multiple evolutionary mechanisms drive papillomavirus diversification". Molecular Biology and Evolution. 24 (5): 1242–58. doi:10.1093/molbev/msm039. PMID 17344207.
- ^ Campo MS, ed. (2006). Papillomavirus Research: From Natural History To Vaccines and Beyond. Caister Academic Press. ISBN 978-1-904455-04-2. [1].
- ^ Bravo IG, de Sanjosé Llongueras S, Gottschling M (2010). "The clinical importance of knowledge about papillomavirus evolution". Trends in Microbiology. 18 (10): 432–8. doi:10.1016/j.tim.2010.07.008. PMID 20739182.
- ^ Rector A, Van Ranst M (October 2013). "Animal papillomaviruses". Virology. 445 (1–2): 213–23. doi:10.1016/j.virol.2013.05.007. PMID 23711385.
- ^ López-Bueno A, Mavian C, Labella AM, Castro D, Borrego JJ, Alcami A, Alejo A (October 2016). "Concurrence of Iridovirus, Polyomavirus, and a Unique Member of a New Group of Fish Papillomaviruses in Lymphocystis Disease-Affected Gilthead Sea Bream". Journal of Virology. 90 (19): 8768–79. doi:10.1128/JVI.01369-16. PMC 5021401. PMID 27440877.
- ^ "Virus Taxonomy: 2024 Release". International Committee on Taxonomy of Viruses. Retrieved 5 March 2025.
- ^ a b Chouhy D, Bolatti EM, Pérez GR, Giri AA (November 2013). "Analysis of the genetic diversity and phylogenetic relationships of putative human papillomavirus types". The Journal of General Virology. 94 (Pt 11): 2480–8. doi:10.1099/vir.0.055137-0. hdl:2133/9862. PMID 23997181.
- ^ "Human papillomavirus reference clones". International Human Papillomavirus Reference Center. Karolinska Institutet. 23 October 2014. Archived from the original on 29 October 2014.
- ^ Van Doorslaer, Koenraad; Chen, Zigui; Bernard, Hans-Ulrich; Chan, Paul K. S.; Desalle, Rob; Dillner, Joakim; Forslund, Ola; Haga, Takeshi; McBride, Alison A.; Villa, Luisa L.; Burk, Robert D. (2018). "ICTV Virus Taxonomy Profile: Papillomaviridae". Journal of General Virology. 99 (8): 989–990. doi:10.1099/jgv.0.001105. PMC 6171710. PMID 29927370.
- ^ a b Regney, Melanie; Kraberger, Simona; Custer, Joy M.; Crane, Adele E.; Shero, Michelle R.; Beltran, Roxanne S.; Kirkham, Amy L.; Van Doorslaer, Koenraad; Stone, Anne C.; Goebel, Michael E.; Burns, Jennifer M.; Varsani, Arvind (2024). "Diverse papillomaviruses identified from Antarctic fur seals, leopard seals and Weddell seals from the Antarctic". Virology. 594. doi:10.1016/j.virol.2024.110064. PMID 38522135.
- ^ Antonsson A, Hansson BG (December 2002). "Healthy skin of many animal species harbors papillomaviruses which are closely related to their human counterparts". Journal of Virology. 76 (24): 12537–42. doi:10.1128/JVI.76.24.12537-12542.2002. PMC 136724. PMID 12438579.
- ^ Holliday, Chuck. "Prof. Chuck Holliday's www page at Lafayette College » Jackalopes". Archived from the original on 2014-07-18. Retrieved 2014-07-13.
- ^ Christensen ND (2005). "Cottontail rabbit papillomavirus (CRPV) model system to test antiviral and immunotherapeutic strategies". Antiviral Chemistry & Chemotherapy. 16 (6): 355–62. doi:10.1177/095632020501600602. PMID 16331841.
- ^ Coggins LW, Ma JQ, Slater AA, Campo MS (June 1985). "Sequence homologies between bovine papillomavirus genomes mapped by a novel low-stringency heteroduplex method". Virology. 143 (2): 603–11. doi:10.1016/0042-6822(85)90398-8. PMID 2998027.
- ^ Van Ranst M, Tachezy R, Pruss J, Burk RD (June 1992). "Primary structure of the E6 protein of Micromys minutus papillomavirus and Mastomys natalensis papillomavirus". Nucleic Acids Research. 20 (11): 2889. doi:10.1093/nar/20.11.2889. PMC 336941. PMID 1319576.
- ^ Varsani A, Kraberger S, Jennings S, Porzig EL, Julian L, Massaro M, Pollard A, Ballard G, Ainley DG (June 2014). "A novel papillomavirus in Adélie penguin (Pygoscelis adeliae) faeces sampled at the Cape Crozier colony, Antarctica". The Journal of General Virology. 95 (Pt 6): 1352–65. doi:10.1099/vir.0.064436-0. PMID 24686913. S2CID 206218507.
- ^ Gottschling M, Göker M, Stamatakis A, Bininda-Emonds OR, Nindl I, Bravo IG (July 2011). "Quantifying the phylodynamic forces driving papillomavirus evolution". Molecular Biology and Evolution. 28 (7): 2101–13. doi:10.1093/molbev/msr030. PMID 21285031.
- ^ Ho L, Chan SY, Burk RD, Das BC, Fujinaga K, Icenogle JP, Kahn T, Kiviat N, Lancaster W, Mavromara-Nazos P (November 1993). "The genetic drift of human papillomavirus type 16 is a means of reconstructing prehistoric viral spread and the movement of ancient human populations". Journal of Virology. 67 (11): 6413–23. doi:10.1128/JVI.67.11.6413-6423.1993. PMC 238076. PMID 8411343.
- ^ a b Calleja-Macias IE, Villa LL, Prado JC, Kalantari M, Allan B, Williamson AL, Chung LP, Collins RJ, Zuna RE, Dunn ST, Chu TY, Cubie HA, Cuschieri K, von Knebel-Doeberitz M, Martins CR, Sanchez GI, Bosch FX, Munoz N, Bernard HU (November 2005). "Worldwide genomic diversity of the high-risk human papillomavirus types 31, 35, 52, and 58, four close relatives of human papillomavirus type 16". Journal of Virology. 79 (21): 13630–40. doi:10.1128/JVI.79.21.13630-13640.2005. PMC 1262609. PMID 16227283.
- ^ Gottschling M, Göker M, Köhler A, Lehmann MD, Stockfleth E, Nindl I (2009). "Cutaneotropic β-/γ-HPV types are rarely shared between family members". Journal of Investigative Dermatology. 129 (10): 2427–34. doi:10.1038/jid.2009.94. PMID 19516265.
- ^ Van Ranst M, Fuse A, Fiten P, Beuken E, Pfister H, Burk RD, Opdenakker G (October 1992). "Human papillomavirus type 13 and pygmy chimpanzee papillomavirus type 1: comparison of the genome organizations". Virology. 190 (2): 587–96. doi:10.1016/0042-6822(92)90896-W. PMID 1325697.
- ^ Willemsen A, Bravo IG (2019) Origin and evolution of papillomavirus (onco)genes and genomes. Philos Trans R Soc Lond B Biol Sci. 374(1773):20180303
- ^ Murahwa AT, Nindo F, Onywera H, Meiring TL, Martin DP, Williamson AL (2019) Evolutionary dynamics of ten novel Gamma-PVs: insights from phylogenetic incongruence, recombination and phylodynamic analyses. BMC Genomics 20(1):368
- ^ Rayment I, Baker TS, Caspar DL, Murakami WT (January 1982). "Polyoma virus capsid structure at 22.5 A resolution". Nature. 295 (5845): 110–5. Bibcode:1982Natur.295..110R. doi:10.1038/295110a0. PMC 4144041. PMID 6276752.
- ^ Joyce JG, Tung JS, Przysiecki CT, Cook JC, Lehman ED, Sands JA, Jansen KU, Keller PM (February 1999). "The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes". The Journal of Biological Chemistry. 274 (9): 5810–22. doi:10.1074/jbc.274.9.5810. PMID 10026203.
- ^ Giroglou T, Florin L, Schäfer F, Streeck RE, Sapp M (February 2001). "Human papillomavirus infection requires cell surface heparan sulfate". Journal of Virology. 75 (3): 1565–70. doi:10.1128/JVI.75.3.1565-1570.2001. PMC 114064. PMID 11152531.
- ^ Evander M, Frazer IH, Payne E, Qi YM, Hengst K, McMillan NA (March 1997). "Identification of the alpha6 integrin as a candidate receptor for papillomaviruses". Journal of Virology. 71 (3): 2449–56. doi:10.1128/JVI.71.3.2449-2456.1997. PMC 191355. PMID 9032382.
- ^ McMillan NA, Payne E, Frazer IH, Evander M (September 1999). "Expression of the alpha6 integrin confers papillomavirus binding upon receptor-negative B-cells". Virology. 261 (2): 271–9. doi:10.1006/viro.1999.9825. PMID 10497112.
- ^ Selinka HC, Giroglou T, Sapp M (August 2002). "Analysis of the infectious entry pathway of human papillomavirus type 33 pseudovirions". Virology. 299 (2): 279–287. doi:10.1006/viro.2001.1493. PMID 12202231.
- ^ Day PM, Lowy DR, Schiller JT (March 2003). "Papillomaviruses infect cells via a clathrin-dependent pathway". Virology. 307 (1): 1–11. doi:10.1016/S0042-6822(02)00143-5. PMID 12667809.
- ^ a b Kämper N, Day PM, Nowak T, Selinka HC, Florin L, Bolscher J, Hilbig L, Schiller JT, Sapp M (January 2006). "A membrane-destabilizing peptide in capsid protein L2 is required for egress of papillomavirus genomes from endosomes". Journal of Virology. 80 (2): 759–68. doi:10.1128/JVI.80.2.759-768.2006. PMC 1346844. PMID 16378978.
- ^ a b Day PM, Baker CC, Lowy DR, Schiller JT (September 2004). "Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression". Proceedings of the National Academy of Sciences of the United States of America. 101 (39): 14252–7. Bibcode:2004PNAS..10114252D. doi:10.1073/pnas.0404229101. PMC 521143. PMID 15383670.
- ^ Zhang, Pengwei; Monteiro Da Silva, Gabriel; Deatherage, Catherine; Burd, Christopher; Dimaio, Daniel (2018). "Cell-Penetrating Peptide Mediates Intracellular Membrane Passage of Human Papillomavirus L2 Protein to Trigger Retrograde Trafficking". Cell. 174 (6): 1465–1476.e13. doi:10.1016/j.cell.2018.07.031. PMC 6128760. PMID 30122350.
- ^ a b Alison A. McBride (18 March 2017). "Mechanisms and strategies of papillomavirus replication". Biological Chemistry. 398 (8): 919–927. doi:10.1515/HSZ-2017-0113. ISSN 1431-6730. PMID 28315855. Wikidata Q39186071.
- ^ Do HT, Koriyama C, Khan NA, Higashi M, Kato T, Le NT, Matsushita S, Kanekura T, Akiba S (January 2013). "The etiologic role of human papillomavirus in penile cancers: a study in Vietnam". British Journal of Cancer. 108 (1): 229–33. doi:10.1038/bjc.2012.583. PMC 3553541. PMID 23299525.
- ^ Gogilashvili K, Shonia N, Burkadze G (December 2012). "The role of human papillomavirus in oral squamous cell carcinoma (review)". Georgian Medical News (213): 32–6. PMID 23293230.
- ^ Preti M, Rotondo JC, Holzinger D, Micheletti L, Gallio N, McKay-Chopin S, Carreira C, Privitera SS, Watanabe R, Ridder R, Pawlita M, Benedetto C, Tommasino M, Gheit T (2020). "Role of Human Papillomavirus Infection in the Etiology of Vulvar Cancer in Italian Women". Infect Agents Cancer. 20: 20. doi:10.1186/s13027-020-00286-8. PMC 7110671. PMID 32266002.
- ^ Tolstov Y, Hadaschik B, Pahernik S, Hohenfellner M, Duensing S (January 2014). "Human papillomaviruses in urological malignancies: a critical assessment". Urologic Oncology. 32 (1): 46.e19–27. doi:10.1016/j.urolonc.2013.06.012. PMID 24140249.
- ^ "PDB101: Molecule of the Month: Human Papillomavirus and Vaccines". RCSB: PDB-101. Retrieved 2018-05-14.
- ^ Guan J, Bywaters SM, Brendle SA, Ashley RE, Makhov AM, Conway JF, Christensen ND, Hafenstein S (6 December 2017). "High-Resolution Structure Analysis of Antibody V5 Conformational Epitope on Human Papillomavirus 16". Viruses. 9 (12): 374. doi:10.3390/v9120374. PMC 5744149. PMID 29211035.
- ^ Meyers C, Frattini MG, Hudson JB, Laimins LA (August 1992). "Biosynthesis of human papillomavirus from a continuous cell line upon epithelial differentiation". Science. 257 (5072): 971–3. Bibcode:1992Sci...257..971M. doi:10.1126/science.1323879. PMID 1323879.
- ^ McLaughlin-Drubin ME, Christensen ND, Meyers C (May 2004). "Propagation, infection, and neutralization of authentic HPV16 virus". Virology. 322 (2): 213–9. doi:10.1016/j.virol.2004.02.011. PMID 15110519.
- ^ Goodsell, D.S (2018). "Human Papillomavirus and Vaccines". RCSB Protein Data Bank. doi:10.2210/rcsb_pdb/mom_2018_5.
- ^ a b c Zheng ZM, Baker CC (September 2006). "Papillomavirus genome structure, expression, and post-transcriptional regulation". Frontiers in Bioscience. 11: 2286–302. doi:10.2741/1971. PMC 1472295. PMID 16720315.
- ^ a b Tang S, Tao M, McCoy JP, Zheng ZM (May 2006). "The E7 oncoprotein is translated from spliced E6*I transcripts in high-risk human papillomavirus type 16- or type 18-positive cervical cancer cell lines via translation reinitiation". Journal of Virology. 80 (9): 4249–63. doi:10.1128/JVI.80.9.4249-4263.2006. PMC 1472016. PMID 16611884.
- ^ Wang X, Meyers C, Wang HK, Chow LT, Zheng ZM (August 2011). "Construction of a full transcription map of human papillomavirus type 18 during productive viral infection". Journal of Virology. 85 (16): 8080–92. doi:10.1128/JVI.00670-11. PMC 3147953. PMID 21680515.
- ^ Jia R, Liu X, Tao M, Kruhlak M, Guo M, Meyers C, Baker CC, Zheng ZM (January 2009). "Control of the papillomavirus early-to-late switch by differentially expressed SRp20". Journal of Virology. 83 (1): 167–80. doi:10.1128/JVI.01719-08. PMC 2612334. PMID 18945760.
- ^ McBride AA, McPhillips MG, Oliveira JG (December 2004). "Brd4: tethering, segregation and beyond". Trends in Microbiology. 12 (12): 527–9. doi:10.1016/j.tim.2004.10.002. PMID 15539109.
- ^ Doorbar J, Campbell D, Grand RJ, Gallimore PH (February 1986). "Identification of the human papilloma virus-1a E4 gene products". The EMBO Journal. 5 (2): 355–62. doi:10.1002/j.1460-2075.1986.tb04219.x. PMC 1166739. PMID 3011404.
- ^ Bravo IG, Alonso A (December 2004). "Mucosal human papillomaviruses encode four different E5 proteins whose chemistry and phylogeny correlate with malignant or benign growth". Journal of Virology. 78 (24): 13613–26. doi:10.1128/JVI.78.24.13613-13626.2004. PMC 533923. PMID 15564472.
- ^ "PDB 2I0I".
- ^ a b c Gupta S, Takhar PP, Degenkolbe R, Koh CH, Zimmermann H, Yang CM, Guan Sim K, Hsu SI, Bernard HU (December 2003). "The human papillomavirus type 11 and 16 E6 proteins modulate the cell-cycle regulator and transcription cofactor TRIP-Br1". Virology. 317 (1): 155–64. doi:10.1016/j.virol.2003.08.008. PMID 14675634.
- ^ a b c Glaunsinger BA, Lee SS, Thomas M, Banks L, Javier R (November 2000). "Interactions of the PDZ-protein MAGI-1 with adenovirus E4-ORF1 and high-risk papillomavirus E6 oncoproteins". Oncogene. 19 (46): 5270–80. doi:10.1038/sj.onc.1203906. PMC 3072458. PMID 11077444.
- ^ "iHOP information Hyperlinked over Proteins UBE3A". Archived from the original on 2007-09-27. Retrieved 2007-05-01.
- ^ "Biochemistry, Nottingham University – 3.0 Enzymes of the Ubiquitin Pathway". Archived from the original on 2007-05-06. Retrieved 2007-05-01.
- ^ Kelley ML, Keiger KE, Lee CJ, Huibregtse JM (March 2005). "The global transcriptional effects of the human papillomavirus E6 protein in cervical carcinoma cell lines are mediated by the E6AP ubiquitin ligase". Journal of Virology. 79 (6): 3737–47. doi:10.1128/JVI.79.6.3737-3747.2005. PMC 1075713. PMID 15731267.
- ^ a b Kiyono T, Hiraiwa A, Fujita M, Hayashi Y, Akiyama T, Ishibashi M (October 1997). "Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the Drosophila discs large tumor suppressor protein". Proceedings of the National Academy of Sciences of the United States of America. 94 (21): 11612–6. Bibcode:1997PNAS...9411612K. doi:10.1073/pnas.94.21.11612. PMC 23554. PMID 9326658.
- ^ Nishimura A, Nakahara T, Ueno T, Sasaki K, Yoshida S, Kyo S, Howley PM, Sakai H (April 2006). "Requirement of E7 oncoprotein for viability of HeLa cells". Microbes and Infection. 8 (4): 984–93. doi:10.1016/j.micinf.2005.10.015. PMID 16500131.
- ^ Jackson ME, Pennie WD, McCaffery RE, Smith KT, Grindlay GJ, Campo MS (1991). "The B subgroup bovine papillomaviruses lack an identifiable E6 open reading frame". Molecular Carcinogenesis. 4 (5): 382–7. doi:10.1002/mc.2940040510. PMID 1654923. S2CID 22514962.
- ^ Carter JJ, Wipf GC, Madeleine MM, Schwartz SM, Koutsky LA, Galloway DA (May 2006). "Identification of human papillomavirus type 16 L1 surface loops required for neutralization by human sera". Journal of Virology. 80 (10): 4664–72. doi:10.1128/JVI.80.10.4664-4672.2006. PMC 1472072. PMID 16641259.
- ^ Campos SK, Ozbun MA (2009). Papavasiliou N (ed.). "Two highly conserved cysteine residues in HPV16 L2 form an intramolecular disulfide bond and are critical for infectivity in human keratinocytes". PLOS ONE. 4 (2) e4463. Bibcode:2009PLoSO...4.4463C. doi:10.1371/journal.pone.0004463. PMC 2636891. PMID 19214230.
- ^ Richards RM, Lowy DR, Schiller JT, Day PM (January 2006). "Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection". Proceedings of the National Academy of Sciences of the United States of America. 103 (5): 1522–7. Bibcode:2006PNAS..103.1522R. doi:10.1073/pnas.0508815103. PMC 1360554. PMID 16432208.
- ^ Yang R, Yutzy WH, Viscidi RP, Roden RB (April 2003). "Interaction of L2 with beta-actin directs intracellular transport of papillomavirus and infection". The Journal of Biological Chemistry. 278 (14): 12546–53. doi:10.1074/jbc.M208691200. PMID 12560332.
- ^ Bossis I, Roden RB, Gambhira R, Yang R, Tagaya M, Howley PM, Meneses PI (June 2005). "Interaction of tSNARE syntaxin 18 with the papillomavirus minor capsid protein mediates infection". Journal of Virology. 79 (11): 6723–31. doi:10.1128/JVI.79.11.6723-6731.2005. PMC 1112158. PMID 15890910.
- ^ Pastrana DV, Gambhira R, Buck CB, Pang YY, Thompson CD, Culp TD, Christensen ND, Lowy DR, Schiller JT, Roden RB (July 2005). "Cross-neutralization of cutaneous and mucosal Papillomavirus types with anti-sera to the amino terminus of L2". Virology. 337 (2): 365–72. doi:10.1016/j.virol.2005.04.011. PMID 15885736.
External links
[edit]- ICTV Report Papillomaviridae
- Viralzone: Papillomaviridae
- Los Alamos National Laboratory maintains a comprehensive (albeit somewhat dated) papillomavirus sequence database. This useful database provides detailed descriptions and references for various papillomavirus types.
- A short video which shows the effects of papillomavirus on the skin of an Indonesian man with epidermodysplasia verruciformis, the genetic inability to defend against some types of cutaneous HPV.
- Best Joint Supplement That Actually Works for Men, Women and Knee de Villiers, E.M., Bernard, H.U., Broker, T., Delius, H. and zur Hausen, H. Index of Viruses – Papillomaviridae (2006). In: ICTVdB – The Universal Virus Database, version 4. Büchen-Osmond, C (Ed), Columbia University, New York, USA.
- 00.099. Papillomaviridae description In: ICTVdB – The Universal Virus Database, version 4. Büchen-Osmond, C. (Ed), Columbia University, New York, USA
- Human papillomavirus particle and genome visualization
- ICTV
| Authority control databases: National |
|---|