
Community hub
Recent from talks
Contribute something
Nothing was collected or created yet.
Racetam
View on Wikipedia

Racetams, also sometimes known simply as pyrrolidones,[1] are a class of drugs that share a pyrrolidone nucleus.[2][3][4] Many, but not all, specifically have a 2-oxo-1-pyrrolidine acetamide (piracetam) nucleus. Some racetams, such as piracetam, aniracetam, oxiracetam, pramiracetam, and phenylpiracetam, are considered nootropics.[5] Phenylpiracetam is also a stimulant.[2] Others, such as levetiracetam, brivaracetam, and seletracetam, are anticonvulsants.[6]
Mechanism
[edit]There is no universally accepted mechanism of action for racetams. Racetams generally show negligible affinity for common central nervous system receptors, but modulation of central neurotransmitters, including acetylcholine and glutamate, has been reported. Although aniracetam and nebracetam show some affinity for muscarinic acetylcholine receptors, only nefiracetam demonstrates nanomolar interactions with neurotransmitter receptors. Modification of membrane-located mechanisms of central signal transduction is another hypothesis.[3]
Like some ampakines, some racetams, such as piracetam and aniracetam, are positive allosteric modulators of the AMPA receptor.[7]
Racetams are understood to work by allosterically modulating glutamate receptors, specifically AMPA receptors, leading to Ca2+ influx that is excitatory.[8] Racetams are posited to enhance memory through interaction with glutamate receptors in the central nervous system.
Phenylpiracetam, unique among racetams in being a phenethylamine and stimulant, is an atypical dopamine reuptake inhibitor.[9][10][11][12]
Anticonvulsant racetams, including levetiracetam, brivaracetam, and seletracetam, act as synaptic vesicle glycoprotein 2A (SV2A) ligands.[6] A newer analogue of these agents, padsevonil, is no longer a racetam itself but is much more potent in comparison and interacts with not only SV2A but also synaptic vesicle glycoprotein 2B (SV2B) and synaptic vesicle glycoprotein 2C (SV2C).[6]
Methylphenylpiracetam, a derivative of phenylpiracetam, is a positive allosteric modulator of the sigma σ1 receptor.[10][13][14][15] It is currently the only racetam known to possess this action.[10]
Cofactors
[edit]In studies with aged rats, marked improvement has been observed in cognitive tasks in experimental groups given piracetam. Performance was further increased when piracetam was combined with choline. Evidence in studies with rats has indicated that the potency of piracetam is increased when administered with choline.[16]
List of racetams
[edit]| Structure | Name |
|---|---|
|
Piracetam |
 |
Oxiracetam |
 |
Phenylpiracetam / Fonturacetam |
 |
Phenylpiracetam Hydrazide / Fonturacetam Hydrazide |
 |
Aniracetam |
 |
Pramiracetam |
 |
Seletracetam |
 |
Levetiracetam |
 |
Coluracetam / BCI-540 |
 |
Fasoracetam |
 |
Brivaracetam |
 |
Dimiracetam |
 |
Methylphenylpiracetam / E1R |
 |
Nebracetam |
 |
Nefiracetam |
 |
Omberacetam / Noopept |
 |
Rolziracetam |
 |
Cebaracetam |
Chemistry
[edit]Racetams are 2-pyrrolidone derivatives and may sometimes be referred to simply as pyrrolidones (2-oxopyrrolidines).[1] Many, but not all, specifically have a 2-oxo-1-pyrrolidine acetamide nucleus, which is the chemical structure of piracetam.
Racetams are cyclic derivatives of the inhibitory neurotransmitter γ-aminobutyric acid (GABA).[2] They are also structurally related to the endogenous cyclic amino acid pyroglutamic acid (pyroglutamate), a cyclic analogue of the endogenous excitatory neurotransmitter glutamic acid (glutamate).[3][4]
-
 γ-Aminobutyric acid (GABA)
γ-Aminobutyric acid (GABA) -

-
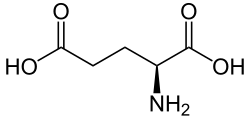 Glutamic acid (glutamate)
Glutamic acid (glutamate) -

-



-Pyroglutamic_acid_Structural_Formulae.png)
Some agents included in the racetam family are not technically racetams themselves in terms of chemical structure and instead are closely related compounds.[17] They may be referred to as "racetam-like".[17][2] These agents include aloracetam, molracetam, omberacetam (noopept), padsevonil, and tenilsetam.[17][6][18]
Society and culture
[edit]Legality
[edit]Australia
[edit]All racetams are schedule 4 substances in Australia under the Poisons Standard (February 2020).[19] A schedule 4 substance is classified as "Prescription Only Medicine, or Prescription Animal Remedy – Substances, the use or supply of which should be by or on the order of persons permitted by State or Territory legislation to prescribe and should be available from a pharmacist on prescription."[19]
References
[edit]- ^ a b Shorvon S (December 2001). "Pyrrolidone derivatives". Lancet. 358 (9296): 1885–1892. doi:10.1016/S0140-6736(01)06890-8. PMID 11741647.
- ^ a b c d Malykh AG, Sadaie MR (February 2010). "Piracetam and piracetam-like drugs: from basic science to novel clinical applications to CNS disorders". Drugs. 70 (3): 287–312. doi:10.2165/11319230-000000000-00000. PMID 20166767.
- ^ a b c Gualtieri F, Manetti D, Romanelli MN, Ghelardini C (2002). "Design and study of piracetam-like nootropics, controversial members of the problematic class of cognition-enhancing drugs". Current Pharmaceutical Design. 8 (2): 125–138. doi:10.2174/1381612023396582. PMID 11812254.
- ^ a b Gouliaev AH, Senning A (May 1994). "Piracetam and other structurally related nootropics". Brain Res Brain Res Rev. 19 (2): 180–222. doi:10.1016/0165-0173(94)90011-6. PMID 8061686.
- ^ Cohen, Pieter A.; Zakharevich, Igor; Gerona, Roy (25 November 2019). "Presence of Piracetam in Cognitive Enhancement Dietary Supplements". JAMA Internal Medicine. 180 (3): 458–459. doi:10.1001/jamainternmed.2019.5507. PMC 6902196. PMID 31764936.
- ^ a b c d Wu PP, Cao BR, Tian FY, Gao ZB (May 2024). "Development of SV2A Ligands for Epilepsy Treatment: A Review of Levetiracetam, Brivaracetam, and Padsevonil". Neurosci Bull. 40 (5): 594–608. doi:10.1007/s12264-023-01138-2. PMC 11127901. PMID 37897555.
- ^ Ahmed AH, Oswald RE (March 2010). "Piracetam defines a new binding site for allosteric modulators of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) receptors". Journal of Medicinal Chemistry. 53 (5): 2197–203. doi:10.1021/jm901905j. PMC 2872987. PMID 20163115.
- ^ Copani A, Genazzani AA, Aleppo G, Casabona G, Canonico PL, Scapagnini U, Nicoletti F (April 1992). "Nootropic drugs positively modulate alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-sensitive glutamate receptors in neuronal cultures". Journal of Neurochemistry. 58 (4): 1199–204. doi:10.1111/j.1471-4159.1992.tb11329.x. PMID 1372342.
- ^ Molina-Carballo A, Checa-Ros A, Muñoz-Hoyos A (July 2016). "Treatments and compositions for attention deficit hyperactivity disorder: a patent review". Expert Opin Ther Pat. 26 (7): 799–814. doi:10.1080/13543776.2016.1182989. PMID 27138211.
The racetams have different activities [e.g., phenylpiracetam is a stimulant developed and marketed in Russia, piracetam is a nootropic, and levetiracetam is widely used as an anticonvulsant (Figure 17)].
- ^ a b c Veinberg G, Vavers E, Orlova N, Kuznecovs J, Domracheva I, Vorona M, Zvejniece L, Dambrova M (2015). "Stereochemistry of phenylpiracetam and its methyl derivative: improvement of the pharmacological profile". Chemistry of Heterocyclic Compounds. 51 (7): 601–606. doi:10.1007/s10593-015-1747-9. ISSN 0009-3122.
Phenylpiracetam was originally designed as a nootropic drug for the sustenance and improvement of the physical condition and cognition abilities of Soviet space crews.2 Later, especially during the last decade, phenylpiracetam was introduced into general clinical practice in Russia and in some Eastern European countries. The possible target receptors and mechanisms for the acute activity of this drug remained unclear, until very recently it was found that (R)-phenylpiracetam (5) (MRZ-9547) is a selective dopamine transporter inhibitor that moderately stimulates striatal dopamine release.19
- ^ Sommer S, Danysz W, Russ H, Valastro B, Flik G, Hauber W (December 2014). "The dopamine reuptake inhibitor MRZ-9547 increases progressive ratio responding in rats". The International Journal of Neuropsychopharmacology. 17 (12): 2045–2056. doi:10.1017/S1461145714000996. PMID 24964269.
Here, we tested the effects of MRZ-9547 [...], and its l-enantiomer MRZ-9546 on effort-related decision making in rats. The racemic form of these compounds referred to as phenotropil has been shown to stimulate motor activity in rats (Zvejniece et al., 2011) and enhance physical capacity and cognition in humans (Malykh and Sadaie, 2010). [...] MRZ-9547 turned out to be a DAT inhibitor as shown by displacement of binding of [125I] RTI-55 (IC50 = 4.82 ± 0.05 μM, n=3) to human recombinant DAT expressed in CHO-K1 cells and inhibition of DA uptake (IC50 = 14.5 ± 1.6 μM, n=2) in functional assays in the same cells. It inhibited norepinephrine transporter (NET) with an IC50 of 182 μM (one experiment in duplicate). The potencies for the l-enantiomer MRZ-9546 were as follows: DAT binding (Ki = 34.8 ± 14.8 μM, n=3), DAT function (IC50 = 65.5 ± 8.3 μM, n=2) and NET function (IC50 = 667 μM, one experiment performed in duplicate).
- ^ Stutz PV, Golani LK, Witkin JM (February 2019). "Animal models of fatigue in major depressive disorder". Physiology & Behavior. 199: 300–305. doi:10.1016/j.physbeh.2018.11.042. PMID 30513290.
In a study performed by Sommer et al. (2014), healthy rats treated with the selective dopamine transport (DAT) inhibitor MRZ-9547 (Fig. 1) chose high effort, high reward more often than their untreated matched controls. Unlike similar studies, however, depressive symptoms were not induced before treatment; rather, baseline healthy controls were compared to healthy rats treated with MRZ-9547. [...] In one study, the selective DAT inhibitor MRZ-9547 increased the number of lever presses more than untreated controls (Sommer et al., 2014). The investigators concluded that such effort-based "decision making in rodents could provide an animal model for motivational dysfunctions related to effort expenditure such as fatigue, e.g. in Parkinson's disease or major depression." Based upon the findings with MRZ-9547, they suggested that this drug mechanism might be a valuable therapeutic entity for fatigue in neurological and neuropsychiatric disorders. [...] A high effort bias been reported with bupropion (Randall et al., 2015), lisdexamfetamine (Yohn etal., 2016e), and the DA uptake blockers MRZ-9547 (Sommer et al., 2014), PRX-14040 (Fig. 1) (Yohn et al., 2016d) and GBR12909 (Fig. 1) (Yohn et al., 2016c).
- ^ Vavers E, Zvejniece L, Maurice T, Dambrova M (2019). "Allosteric Modulators of Sigma-1 Receptor: A Review". Front Pharmacol. 10 223. doi:10.3389/fphar.2019.00223. PMC 6433746. PMID 30941035.
- ^ Zvejniece L, Vavers E, Svalbe B, Vilskersts R, Domracheva I, Vorona M, Veinberg G, Misane I, Stonans I, Kalvinsh I, Dambrova M (February 2014). "The cognition-enhancing activity of E1R, a novel positive allosteric modulator of sigma-1 receptors". Br J Pharmacol. 171 (3): 761–771. doi:10.1111/bph.12506. PMC 3969087. PMID 24490863.
- ^ Vavers E, Zvejniece L, Veinberg G, Svalbe B, Domracheva I, Vilskersts R, Dambrova M (2015). "Novel positive allosteric modulators of sigma-1 receptor". SpringerPlus. 4 (Suppl 1) P51. doi:10.1186/2193-1801-4-S1-P51. PMC 4797911.
The R-configuration enantiomers of methylphenylpiracetam are more active positive allosteric modulators of Sigma-1 receptor than S-configuration enantiomers.
- ^ Bartus RT, Dean RL, Sherman KA, Friedman E, Beer B (1981). "Profound effects of combining choline and piracetam on memory enhancement and cholinergic function in aged rats". Neurobiology of Aging. 2 (2): 105–11. doi:10.1016/0197-4580(81)90007-5. PMID 7301036.
- ^ a b c "Classification Status of Racetams" (PDF). Medsafe. 2015. Retrieved 1 October 2024.
Many of the products are based on a group of compounds known collectively as racetams or 'racetam-like' substances. [...] In addition, three substances that do not strictly meet the racetam definition (due to lack of a 2-pyrrolidone ring) are typically described as being part of the racetam family. These are aloracetam, molracetam and noopept.
- ^ "The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances 2018" (PDF). Archived from the original (PDF) on 2021-09-27.
- ^ a b Poisons Standard February 2020. comlaw.gov.au
Racetam
View on GrokipediaHistory and Development
Discovery and Early Research
Piracetam, the first compound in the racetam class, was synthesized in 1964 by Corneliu E. Giurgea and his team at UCB Pharma, a Belgian pharmaceutical company.[8] The molecule was developed as a derivative of γ-aminobutyric acid (GABA), with the aim of creating a compound capable of crossing the blood-brain barrier to exert central nervous system effects, initially targeted toward anxiolytic or sedative applications.[9] Unlike GABA itself, which does not readily penetrate the brain, piracetam's structure allowed for potential modulation of inhibitory neurotransmission.[9] Initial pharmacological evaluations in the mid-1960s shifted focus from anxiolysis after animal studies demonstrated piracetam's ability to counteract amnesia induced by hypoxia and electroshock, preserving memory consolidation in rodents without producing stimulant-like behavioral activation or sedation.[10] These observations highlighted its protective effects on cognitive function under conditions of brain insult, prompting further investigation into learning and memory enhancement in both animal models and preliminary human trials.[10] Giurgea formalized these properties in 1972 by introducing the term "nootropic," derived from Greek roots meaning "mind-turning," to classify agents like piracetam that selectively enhance higher integrative brain functions such as learning and retrieval, while offering neuroprotection, lacking typical psychotropic side effects, and maintaining low toxicity even at high doses.[7] This conceptualization distinguished nootropics from conventional stimulants or sedatives, emphasizing empirical evidence from piracetam's profile in reversing cognitive deficits without impairing normal performance.[7]Expansion of the Class
Following the synthesis of piracetam in 1964, pharmaceutical companies pursued structural analogs in the 1970s to enhance potency and target cognitive deficits associated with dementia and aging. Oxiracetam, developed in 1974 by the Italian firm Istituto di Chimica Farmaceutica (ICF) under code ISF 2522, incorporated a hydroxyl group on the pyrrolidone ring, aiming for greater central nervous system stimulation and improved memory facilitation compared to piracetam.[11] Aniracetam, synthesized around 1978 by Swiss firm F. Hoffmann-La Roche, featured an anisoyl group addition to increase lipophilicity and bioavailability, with early explorations focusing on its potential to amplify glutamatergic signaling for dementia symptom relief. Pramiracetam, created in the late 1970s by Parke-Davis (a Warner-Lambert division), modified the amide side chain with a dipropan-2-ylaminoethyl group, reportedly yielding 8-30 times the potency of piracetam in animal models of learning impairment.[12] In the Soviet Union, phenylpiracetam emerged in 1983 at the Russian Academy of Sciences, adding a phenyl ring to piracetam's structure to confer stimulant properties for countering spaceflight-related stress in cosmonauts, alongside cognitive restoration in post-ischemic conditions. Initial European and Soviet clinical trials from the 1970s to 1980s, including those on oxiracetam and piracetam analogs, reported modest improvements in stroke recovery metrics such as neurological scores and functional independence, though results varied due to small sample sizes and heterogeneous patient populations.[13] By the 2000s, coluracetam was developed by Japan's Mitsubishi Tanabe Pharma, introducing a coumarin moiety to selectively boost high-affinity choline uptake, with preclinical work targeting Alzheimer's-like cholinergic deficits.[14] Post-1990s regulatory challenges in Western markets, including stringent efficacy requirements for nootropics, shifted many racetams from prescription development to research chemical status, limiting large-scale trials but preserving interest in their empirical utility from earlier European data on dementia stabilization and vascular recovery.[15] These modifications generally sought to optimize pharmacokinetics—such as faster onset or brain penetration—while retaining the pyrrolidone core for presumed neuroprotective effects in hypoxic or degenerative states.[16]Chemical Structure
Core Features
Racetams constitute a class of synthetic compounds characterized by a common 2-oxo-1-pyrrolidine nucleus, comprising a five-membered heterocyclic ring with a lactam functionality at the 2-position.[1] This pyrrolidone core, derived from 2-pyrrolidone, forms the foundational scaffold shared across the family, typically featuring nitrogen substitution with acyl or amide moieties that distinguish specific analogs.[3] For instance, piracetam, the prototypical racetam, incorporates a 2-(2-oxopyrrolidin-1-yl)acetamide structure, where the nitrogen of the ring links to a simple acetamide side chain.[17] Substitutions on the amide or attached groups vary widely, altering molecular properties without disrupting the core ring system; aniracetam, for example, bears a 4-methoxybenzoyl group at the pyrrolidine nitrogen, enhancing its distinction from unsubstituted forms.[18] These modifications influence lipophilicity, with baseline racetams like piracetam displaying high hydrophilicity due to polar groups, resulting in slower blood-brain barrier permeation.[19] In contrast, phenyl-substituted variants such as phenylpiracetam exhibit greater lipid solubility from the aromatic addition, facilitating improved central nervous system entry.[20][21] Most racetams manifest as stable, white crystalline powders with generally favorable water solubility, supporting their formulation for oral use; piracetam, for example, dissolves readily in water at concentrations up to 479 mg/mL.[22][17] However, analogs like aniracetam demonstrate reduced aqueous solubility, necessitating alternative solvents such as ethanol or DMSO for dissolution.[23] These solubility profiles stem directly from the polar nature of the pyrrolidone ring and side chain functionalities, ensuring chemical stability under physiological conditions.[24]Synthesis Methods
The prototypical racetam, piracetam (2-oxo-1-pyrrolidinacetamide), is synthesized through a multi-step process starting from 2-pyrrolidone. In one established route, 2-pyrrolidone is first deprotonated using sodium methoxide to form its sodium salt, which then undergoes acylation with chloroacetyl chloride to produce 2-chloro-1-(2-oxopyrrolidin-1-yl)ethan-1-one. This intermediate is subsequently treated with ammonia to afford piracetam via nucleophilic substitution.[25] This method typically yields piracetam in high purity after purification, with overall efficiencies reported around 80-90% in optimized conditions.[26] 2-Pyrrolidone, the core lactam scaffold common to most racetams, can itself be prepared by cyclodehydration of gamma-aminobutyric acid (GABA) under acidic or thermal conditions, though industrial production often employs alternative routes such as the reaction of butyrolactone with ammonia.[3] Variations in the acylation step allow for the synthesis of other racetams; for instance, aniracetam incorporates an anisoyl group via reaction with p-methoxybenzoyl chloride, while pramiracetam uses a longer-chain amide derived from 2-(2-oxopyrrolidin-1-yl)acetamide extension.[27] For substituted analogs like phenylpiracetam (5-phenyl-2-oxo-1-pyrrolidineacetamide), the synthesis begins with beta-phenyl-GABA (phenibut), which is cyclized under dehydrating conditions to form 5-phenyl-2-pyrrolidone, followed by N-acetylation analogous to piracetam.[28] This approach avoids regioselectivity issues in direct arylation of the pyrrolidone ring, with reported yields for the cyclization step exceeding 70%.[29] Chiral racetams, such as levetiracetam ((S)-2-(2-oxopyrrolidin-1-yl)butanamide), require enantioselective methods to control stereochemistry at the alpha-carbon of the side chain, often involving asymmetric hydrogenation or chiral auxiliaries in the amidation step, which can reduce scalability due to additional purification needs and lower enantiomeric excess in non-optimized routes (typically 70-95% ee).[30] Modern multi-component reactions, like the Ugi four-center three-component condensation using gamma-keto acids, offer streamlined access to diverse racetams but are less common for large-scale production due to byproduct management.[31]Pharmacology
Pharmacokinetics
Racetams are characterized by rapid oral absorption, with peak plasma concentrations typically achieved within 1-3 hours following administration. For piracetam, the prototype compound, absorption is extensive and nearly complete, yielding a bioavailability approaching 100%; a single 1400 mg dose results in a maximum plasma concentration of approximately 84 µg/mL at 1 hour under fasting conditions, though food intake reduces this peak by about 17% and delays it to 1.5 hours.[17] Similar rapid kinetics are observed in oxiracetam, where an 800 mg oral dose produces peak serum levels of 25 ± 6 µg/mL within 1-3 hours.[32] Phenylpiracetam also exhibits complete oral bioavailability of 100%, with maximum blood concentrations reached after 1 hour.[33] In contrast, aniracetam shows rapid gastrointestinal absorption but markedly lower systemic bioavailability of around 0.2%, attributable to extensive first-pass metabolism.[34] Distribution across the class involves ready penetration of the blood-brain barrier, facilitated by their structural features, with piracetam demonstrating a volume of distribution of 0.6 L/kg and no significant plasma protein binding; it accumulates in cerebrospinal fluid, where peak levels lag plasma peaks by several hours (Tmax ~5 hours) and half-life extends to 8.5 hours.[17] Brain tissue concentrations often exceed plasma levels in a dose-dependent manner for several racetams, reflecting selective retention.[5] Metabolism is minimal for most racetams, with piracetam undergoing negligible hepatic transformation and oxiracetam similarly showing limited biotransformation.[17][32] Excretion occurs predominantly via the kidneys as unchanged parent compound; for piracetam, 80-100% of the dose appears in urine, approximately 90% unmodified, while oxiracetam recovers 84% unchanged within 24 hours.[17][32] Elimination half-lives vary, ranging from 3-6 hours for oxiracetam and phenylpiracetam to about 5 hours for piracetam, with no significant accumulation upon repeated dosing in short-term studies.[32][17][33] These profiles indicate linear pharmacokinetics over typical dose ranges, with low intersubject variability.[17]Pharmacodynamics
Racetams primarily modulate ionotropic glutamate receptors, with piracetam binding to a distinct allosteric site at the dimer interface of AMPA receptor subunits GluA2 and GluA3, facilitating receptor dimerization and decreasing rates of desensitization and deactivation to elevate channel opening probability upon glutamate binding, independent of direct agonist effects.[4] This action occurs at low binding occupancy and enhances AMPA receptor-mediated excitatory postsynaptic currents in neuronal preparations.[4] Binding affinities remain modest, with piracetam exhibiting micromolar interactions at these sites rather than high-affinity orthosteric binding typical of agonists.[35] Several racetams indirectly potentiate cholinergic activity by promoting acetylcholine release from presynaptic terminals. Oxiracetam and aniracetam, for example, dose-dependently increase acetylcholine efflux from rat hippocampal slices, with peak effects at concentrations of 10-100 μM, suggesting facilitation of hippocampal cholinergic pathways without direct receptor agonism.[36] Piracetam similarly augments acetylcholine release and elevates cholinergic receptor density in cortical regions, contributing to restored neurotransmission in compromised states.[37][38] In rodent models of focal cerebral ischemia, such as middle cerebral artery occlusion, piracetam reduces infarct volume by up to 30-50% when administered post-ischemia, empirically demonstrating mitigation of glutamate-driven excitotoxicity through AMPA receptor modulation that curbs excessive calcium influx and neuronal damage.[39] These neuroprotective outcomes correlate with doses of 100-400 mg/kg in rats, yielding plasma levels comparable to human therapeutic ranges, and persist across multiple studies without altering baseline hemodynamics.[39][40]List of Notable Racetams
- Piracetam: The prototype racetam, characterized by its core 2-oxo-1-pyrrolidine acetamide structure, has been approved in several European countries for treating cortical myoclonus and studied for vertigo due to its effects on vestibular and oculomotor nuclei.[5][41]
- Aniracetam: Features an anisoyl group attached to the pyrrolidone ring, distinguishing it structurally; research has explored its anxiolytic properties mediated by interactions with cholinergic, dopaminergic, and serotonergic systems in animal models.[42][43]
- Oxiracetam: Incorporates a hydroxy group at the 4-position of the pyrrolidone ring; investigated for cognitive deficits in vascular dementia models, where it ameliorates learning impairments and neuronal damage in rats.[44][45]
- Phenylpiracetam (also known as fonturacetam): Modified by addition of a phenyl group at the 4-position of the pyrrolidone ring, enhancing blood-brain barrier penetration; developed for stimulant-like effects and used in Russian cosmonaut programs to counter stress and fatigue during space missions.[46][47]
- Pramiracetam: Distinguished by a dipropyl-substituted amide chain; noted for increasing high-affinity choline uptake (HACU) in hippocampal synaptosomes, supporting cholinergic enhancement in preclinical studies.[48][49]
- Coluracetam: Features a structural variation targeting high-affinity choline uptake enhancement; preclinical data indicate potential in cholinergic hypofunction without primary effects on serotonin reuptake.[50][14]