Recent from talks
Electron configuration
Knowledge base stats:
Talk channels stats:
Members stats:
Electron configuration
In atomic physics and quantum chemistry, the electron configuration is the distribution of electrons of an atom or molecule (or other physical structure) in atomic or molecular orbitals. For example, the electron configuration of the neon atom is 1s2 2s2 2p6, meaning that the 1s, 2s, and 2p subshells are occupied by two, two, and six electrons, respectively.
Electronic configurations describe each electron as moving independently in an orbital, in an average field created by the nuclei and all the other electrons. Mathematically, configurations are described by Slater determinants or configuration state functions.
According to the laws of quantum mechanics, a level of energy is associated with each electron configuration. In certain conditions, electrons are able to move from one configuration to another by the emission or absorption of a quantum of energy, in the form of a photon.
Knowledge of the electron configuration of different atoms is useful in understanding the structure of the periodic table of elements, for describing the chemical bonds that hold atoms together, and in understanding the chemical formulas of compounds and the geometries of molecules. In bulk materials, this same idea helps explain the peculiar properties of lasers and semiconductors.
Electron configuration was first conceived under the Bohr model of the atom, and it is still common to speak of shells and subshells despite the advances in understanding of the quantum-mechanical nature of electrons.
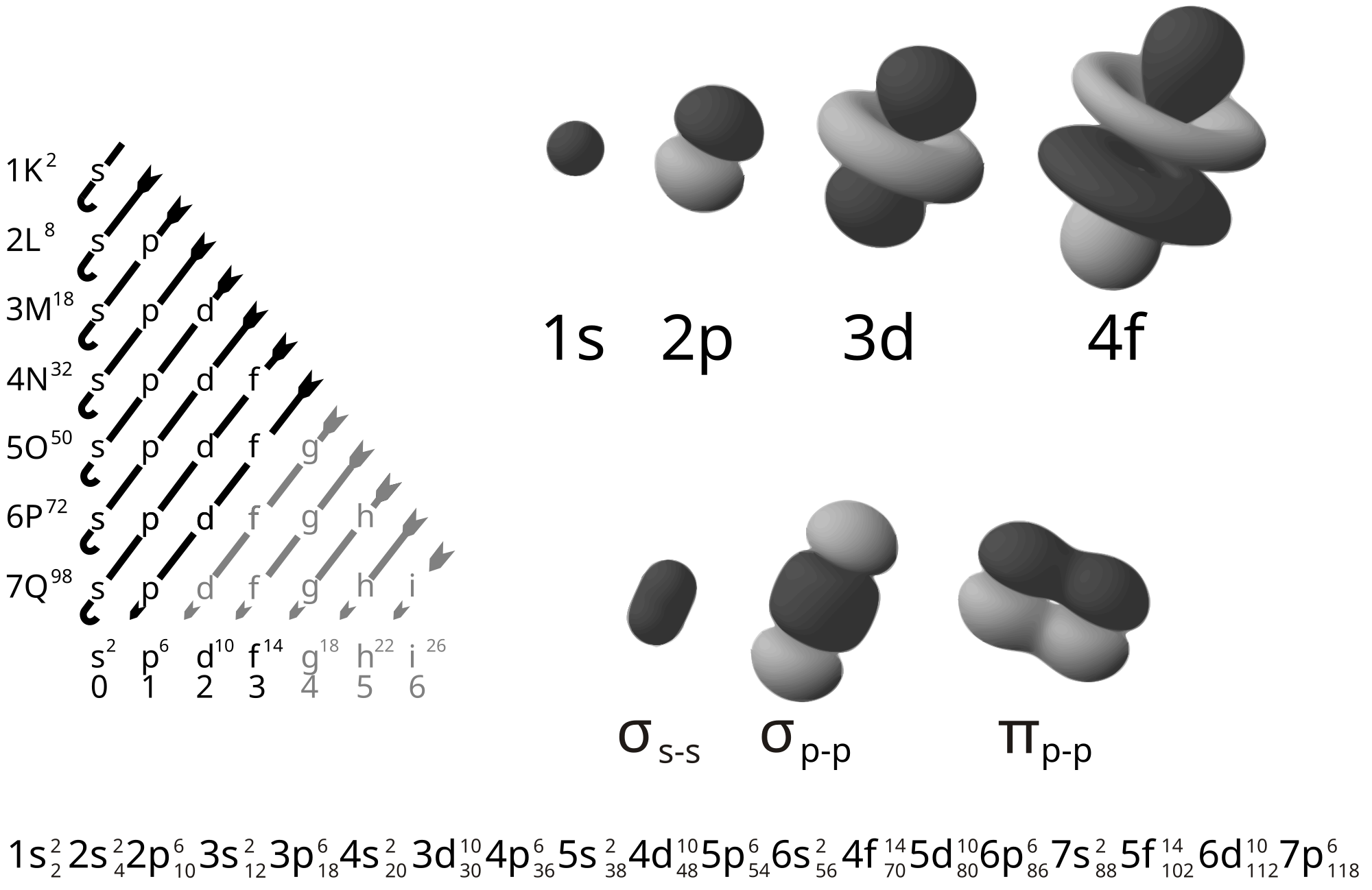
An electron shell is the set of allowed states that share the same principal quantum number, n, that electrons may occupy. In each term of an electron configuration, n is the positive integer that precedes each orbital letter (e.g. helium's electron configuration is 1s2, therefore n = 1, and the orbital contains two electrons). An atom's nth electron shell can accommodate 2n2 electrons. For example, the first shell can accommodate two electrons, the second shell eight electrons, the third shell eighteen, and so on. The factor of two arises because the number of allowed states doubles with each successive shell due to electron spin—each atomic orbital admits up to two otherwise identical electrons with opposite spin, one with a spin +1⁄2 (usually denoted by an up-arrow) and one with a spin of −1⁄2 (with a down-arrow).
A subshell is the set of states defined by a common azimuthal quantum number, l, within a shell. The value of l is in the range from 0 to n − 1. The values l = 0, 1, 2, 3 correspond to the s, p, d, and f labels, respectively. For example, the 3d subshell has n = 3 and l = 2. The maximum number of electrons that can be placed in a subshell is given by 2(2l + 1). This gives two electrons in an s subshell, six electrons in a p subshell, and ten electrons in a d subshell.
The numbers of electrons that can occupy each shell and each subshell arise from the equations of quantum mechanics, in particular the Pauli exclusion principle, which states that no two electrons in the same atom can have the same values of the four quantum numbers.
Hub AI
Electron configuration AI simulator
(@Electron configuration_simulator)
Electron configuration
In atomic physics and quantum chemistry, the electron configuration is the distribution of electrons of an atom or molecule (or other physical structure) in atomic or molecular orbitals. For example, the electron configuration of the neon atom is 1s2 2s2 2p6, meaning that the 1s, 2s, and 2p subshells are occupied by two, two, and six electrons, respectively.
Electronic configurations describe each electron as moving independently in an orbital, in an average field created by the nuclei and all the other electrons. Mathematically, configurations are described by Slater determinants or configuration state functions.
According to the laws of quantum mechanics, a level of energy is associated with each electron configuration. In certain conditions, electrons are able to move from one configuration to another by the emission or absorption of a quantum of energy, in the form of a photon.
Knowledge of the electron configuration of different atoms is useful in understanding the structure of the periodic table of elements, for describing the chemical bonds that hold atoms together, and in understanding the chemical formulas of compounds and the geometries of molecules. In bulk materials, this same idea helps explain the peculiar properties of lasers and semiconductors.
Electron configuration was first conceived under the Bohr model of the atom, and it is still common to speak of shells and subshells despite the advances in understanding of the quantum-mechanical nature of electrons.
An electron shell is the set of allowed states that share the same principal quantum number, n, that electrons may occupy. In each term of an electron configuration, n is the positive integer that precedes each orbital letter (e.g. helium's electron configuration is 1s2, therefore n = 1, and the orbital contains two electrons). An atom's nth electron shell can accommodate 2n2 electrons. For example, the first shell can accommodate two electrons, the second shell eight electrons, the third shell eighteen, and so on. The factor of two arises because the number of allowed states doubles with each successive shell due to electron spin—each atomic orbital admits up to two otherwise identical electrons with opposite spin, one with a spin +1⁄2 (usually denoted by an up-arrow) and one with a spin of −1⁄2 (with a down-arrow).
A subshell is the set of states defined by a common azimuthal quantum number, l, within a shell. The value of l is in the range from 0 to n − 1. The values l = 0, 1, 2, 3 correspond to the s, p, d, and f labels, respectively. For example, the 3d subshell has n = 3 and l = 2. The maximum number of electrons that can be placed in a subshell is given by 2(2l + 1). This gives two electrons in an s subshell, six electrons in a p subshell, and ten electrons in a d subshell.
The numbers of electrons that can occupy each shell and each subshell arise from the equations of quantum mechanics, in particular the Pauli exclusion principle, which states that no two electrons in the same atom can have the same values of the four quantum numbers.
Recent media